Endocrinology
Key Points
The endocrine system is widespread and consists of glands that exert their effects by means of chemicals (hormones) secreted into the blood circulation (Fig. 6.1). Hormones are chemical messengers of various types, which usually act at some distance from their source, and can be classified according to their main function.
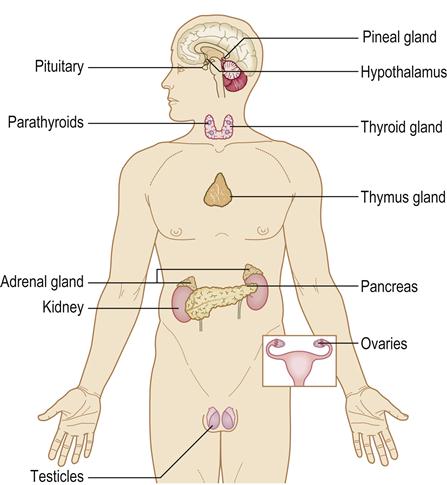
Nervous and endocrine control mechanisms normally maintain body homeostasis, coordinated via the neuroendocrine system and most apparent in the hypothalamus. The hypothalamus, along with the pituitary gland, controls many other endocrine functions.
Pregnancy and the menopause cause important endocrine changes (Ch. 25). It is also common to see endocrine disorders in people who overexercise or who are anorectic, starving, stressed or ill (Table 6.1). Obesity may be due to endocrine disease (e.g. hypothyroidism, insulinoma, Cushing syndrome, polycystic ovary disease).
Table 6.1
Endocrine responses to severe stress or illness
| Plasma levels of hormone rise | Plasma levels of hormone fall | Resistance of hormone increases |
| Adrenaline (epinephrine) | Luteinizing hormone and follicle-stimulating hormone | Insulin |
| Glucagon | Sex steroids | |
| Glucocorticoids and adrenocorticotropic hormone | Thyroid hormones and thyroid-stimulating hormone | |
| Growth hormone | ||
| Prolactin |
Endocrine glands can underproduce (hypofunction) or overproduce (hyperfunction) their hormones. Endocrine disorders (endocrino-pathies) may be caused by diminished hormone release (e.g. hypothyroidism), by hormone resistance (e.g. type 2 diabetes) or by excess (e.g. primary hyperparathyroidism). Endocrinopathies can cause a range of symptoms and signs but fatigue is common to many (e.g. thyroid dysfunction, hyper- or hypoglycaemia, dyslipidaemia and gonadal dysfunction).
Endocrine function can be assessed by measuring the hormone in the blood plasma (many have a circadian rhythm – the level varies over 24 hours), or the hormone or its metabolite in the urine; alternatively, dynamic tests of hormone secretion or regulation may be performed – by suppressing (tests hormone excess) or stimulating (tests hormone deficiency) hormone release; or by assessing levels of hormone receptors or effects on the target tissues.
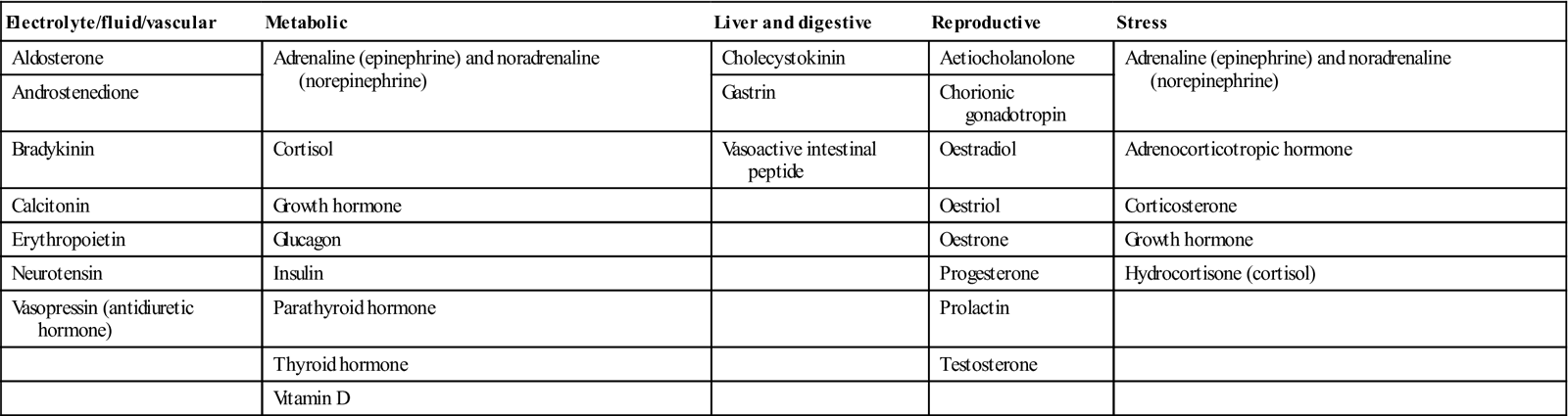
The most common and important specific endocrine disorder is diabetes mellitus, but the hypothalamus is the conductor of the endocrine ‘orchestra’ (Table 6.2).
Table 6.2
Hormones – classified by function
< ?comst?>
| Electrolyte/fluid/vascular | Metabolic | Liver and digestive | Reproductive | Stress |
| Aldosterone | Adrenaline (epinephrine) and noradrenaline (norepinephrine) | Cholecystokinin | Aetiocholanolone | Adrenaline (epinephrine) and noradrenaline (norepinephrine) |
| Androstenedione | Gastrin | Chorionic gonadotropin | ||
| Bradykinin | Cortisol | Vasoactive intestinal peptide | Oestradiol | Adrenocorticotropic hormone |
| Calcitonin | Growth hormone | Oestriol | Corticosterone | |
| Erythropoietin | Glucagon | Oestrone | Growth hormone | |
| Neurotensin | Insulin | Progesterone | Hydrocortisone (cortisol) | |
| Vasopressin (antidiuretic hormone) | Parathyroid hormone | Prolactin | ||
| Thyroid hormone | Testosterone | |||
| Vitamin D |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Hypothalamus and Pituitary Gland
The hypothalamus is the part of the brain that controls many other endocrine glands, via pituitary function and hypophysiotropic hormones sometimes termed ‘releasing factors’.
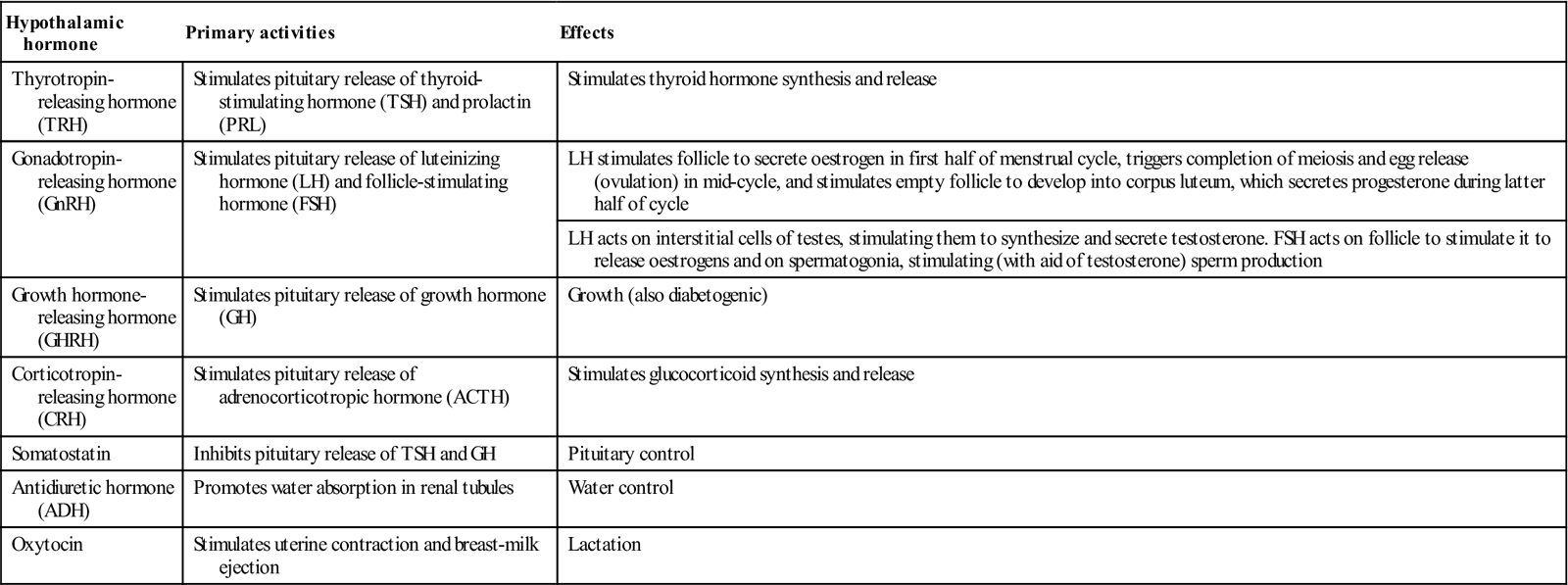
The anterior pituitary (adenohypophysis) originates as an outgrowth from the stomatodeum (Rathke pouch) and, although anatomically distinct from the hypothalamus, falls under its influence by factors passing through a portal venous system (Fig. 6.2). Hypothalamic hormones that act on the anterior pituitary gland include thyrotropin-releasing hormone (TRH), gonadotropin-releasing hormone (GnRH), growth hormone-releasing hormone (GHRH), corticotropin-releasing hormone (CRH) and somatostatin (Table 6.3).
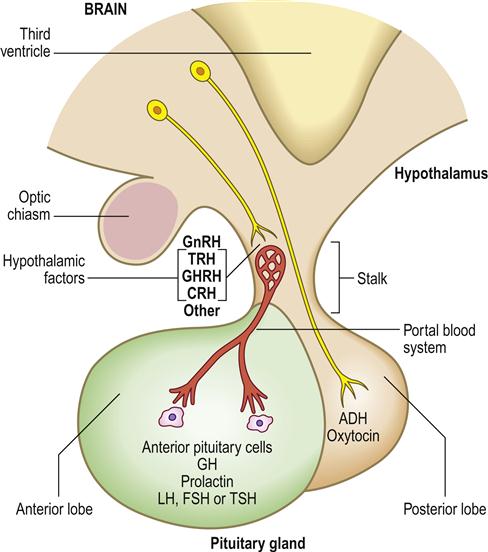
ADH=antidiuretic hormone; CRH=corticotropin-releasing hormone; FSH=follicle stimulating hormone; GH=growth hormone; GHRH=growth hormone-releasing hormone; GnRH=gonadotropin-releasing hormone; LH=luteinizing hormone; TRH=thyrotropin-releasing hormone; TSH=thyroid-stimulating hormone.
Table 6.3
< ?comst?>
| Hypothalamic hormone | Primary activities | Effects |
| Thyrotropin- releasing hormone (TRH) | Stimulates pituitary release of thyroid-stimulating hormone (TSH) and prolactin (PRL) | Stimulates thyroid hormone synthesis and release |
| Gonadotropin- releasing hormone (GnRH) | Stimulates pituitary release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) | LH stimulates follicle to secrete oestrogen in first half of menstrual cycle, triggers completion of meiosis and egg release (ovulation) in mid-cycle, and stimulates empty follicle to develop into corpus luteum, which secretes progesterone during latter half of cycle |
| LH acts on interstitial cells of testes, stimulating them to synthesize and secrete testosterone. FSH acts on follicle to stimulate it to release oestrogens and on spermatogonia, stimulating (with aid of testosterone) sperm production | ||
| Growth hormone- releasing hormone (GHRH) | Stimulates pituitary release of growth hormone (GH) | Growth (also diabetogenic) |
| Corticotropin- releasing hormone (CRH) | Stimulates pituitary release of adrenocorticotropic hormone (ACTH) | Stimulates glucocorticoid synthesis and release |
| Somatostatin | Inhibits pituitary release of TSH and GH | Pituitary control |
| Antidiuretic hormone (ADH) | Promotes water absorption in renal tubules | Water control |
| Oxytocin | Stimulates uterine contraction and breast-milk ejection | Lactation |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
The posterior pituitary (neurohypophysis) is a downgrowth from the base of the brain and is connected with the hypothalamus by neurons. Two hypothalamic hormones act on the posterior pituitary gland – antidiuretic hormone (ADH) and oxytocin.
The hypothalamus is under the control of higher centres in the brain and inhibited by the hormone somatostatin (octreotide). Feedback control influences the amount of hypothalamic hypophysiotropic hormone secreted. Dopamine inhibits pituitary release of prolactin (PRL).
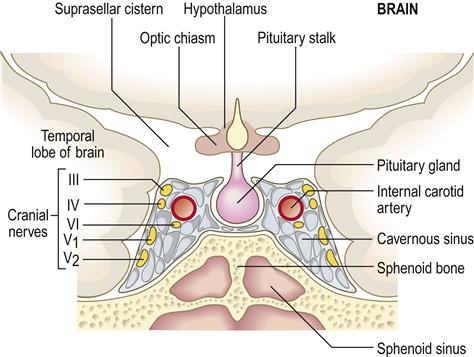
Pituitary hyperfunction is sometimes caused by pituitary tumours, usually microadenomas (smaller than 1 cm), which can have pressure effects on adjacent structures such as the rest of the anterior pituitary gland and the optic chiasm (Fig. 6.3), causing headaches and visual defects as well as effects on hormone release. Around 50% of pituitary adenomas are hormone-secreting – half producing prolactin, the rest producing growth hormone, adrenocorticotropic hormone (ACTH) or thyroid-stimulating hormone (TSH).
Pituitary hormones, used to stimulate growth, have been implicated in the transmission of some cases of iatrogenic Creutzfeldt–Jakob disease (Ch. 13).
Posterior Pituitary Hypofunction
Diabetes insipidus
General aspects
Diabetes insipidus is a rare disease caused by lack of ADH effect. Rarely, this is because of renal insensitivity to ADH (nephrogenic diabetes insipidus – a rare X-linked disorder affecting renal tubular ADH receptors, or due to hypercalcaemia, hypokalaemia, renal disease or drugs such as lithium). More commonly, it is because of reduced ADH secretion – cranial diabetes insipidus, which is caused by head injuries (when it may be temporary), a tumour (usually craniopharyngioma), infiltration or vascular disease in the region of the hypothalamus or pituitary, or it may be idiopathic.
Clinical features
Diabetes insipidus is characterized by an excessive volume of dilute urine (polyuria) and by persistent thirst and drinking (polydipsia). Cranial diabetes insipidus may also cause pressure on the optic chiasm, leading to visual or cranial nerve defects, or raised intracranial pressure and headaches.
General management
The diagnosis is established by assessing the relation of plasma to urine osmolality and by demonstrating an inability to concentrate the urine during a water-deprivation test. The local extent of disease is assessed by imaging (skull radiography and magnetic resonance imaging [MRI]), visual field charting and tests of anterior pituitary function.
Diabetes insipidus is treated with the ADH-like peptide desmopressin or antidiuretic drugs such as chlorpropamide or carbamazepine.
Dental aspects
Local anaesthesia (LA) is the most satisfactory means of pain control. Conscious sedation (CS) may be needed to control anxiety. Dentistry is usually uncomplicated by this disorder except for dryness of the mouth.
Carbamazepine used in the treatment of trigeminal neuralgia may have an additive effect with other drugs used to treat diabetes insipidus. Transient diabetes insipidus can be a complication of head injury, but head injury can also cause the opposite effect – excessive ADH levels.
Syndrome of inappropriate antidiuretic hormone secretion (SIADH)
Excessive ADH levels may be caused by: maxillofacial injury or even elective maxillofacial surgery, head injury or intracranial lesions; general anaesthesia (GA); pain; fits; smoking; tumours (especially some lung cancers); or drugs (carbamazepine, chlorpropamide, vinca alkaloids).
Inappropriate ADH secretion causes water retention, overhydration, confusion, behavioural disturbances, ataxia and dysphagia. Diagnosis is by finding hyponatraemia with concentrated urine and high plasma and urinary ADH, and an abnormal water-excretion test.
Patients with SIADH are treated with fluid restriction, corticosteroids or demeclocycline.
Anterior Pituitary Hypofunction
General aspects
The usual causes of hypopituitarism are local hypothalamic or pituitary lesions, such as tumours, or irradiation (similar factors to those causing cranial diabetes insipidus) or infarction of the pituitary following postpartum haemorrhage (Sheehan syndrome).
Clinical features
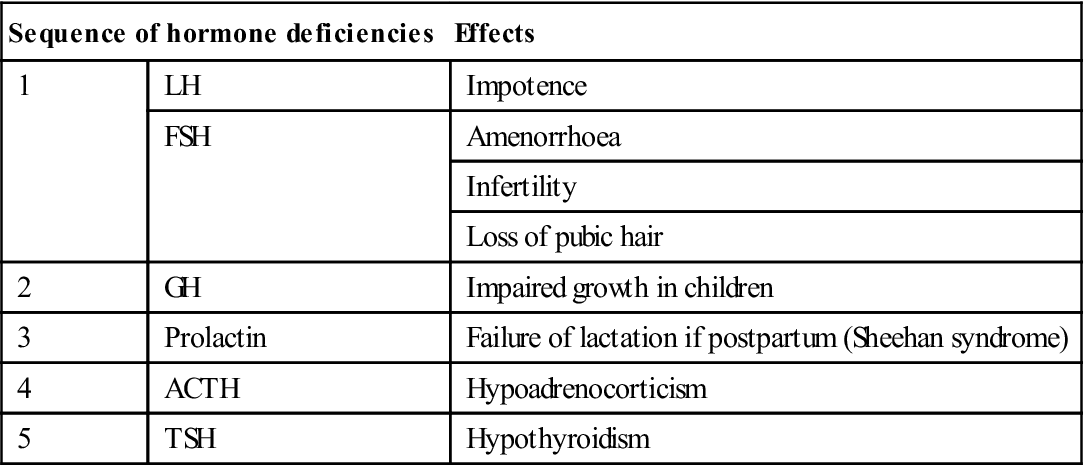
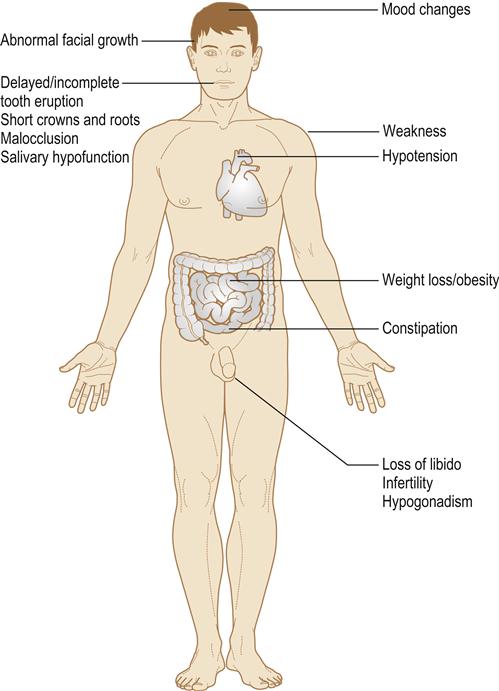
The results of hypopituitarism (Table 6.4) are essentially hypofunction of the target glands (the gonads, thyroid and/or adrenal cortex) with soft skin, hypotension, hypoadrenalism and hypothyroidism.
Table 6.4
Hypopituitarism: clinical effects
< ?comst?>
| Sequence of hormone deficiencies | Effects | |
| 1 | LH | Impotence |
| FSH | Amenorrhoea | |
| Infertility | ||
| Loss of pubic hair | ||
| 2 | GH | Impaired growth in children |
| 3 | Prolactin | Failure of lactation if postpartum (Sheehan syndrome) |
| 4 | ACTH | Hypoadrenocorticism |
| 5 | TSH | Hypothyroidism |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Multiple (panhypopituitarism) hormone deficiencies can result (Fig. 6.4).
General management
Hormone substitution therapy (corticosteroids and thyroxine) is needed. Surgery may be required if there are tumours, cranial nerve defects or hydrocephalus.
Dental aspects
Patients are at risk from adrenal crisis (Ch. 1) and from hypopituitary coma, which is precipitated by stress (trauma, surgery, GA, sedatives or hypnotics, or infection), much in the way that an adrenal crisis may be.
Hypopituitary coma should be treated by laying the patient flat and giving an immediate intravenous injection of 200 mg hydrocortisone sodium succinate. Blood should be taken for assay of glucose, thyroid hormones and cortisol, and 25–50 g dextrose should be given intravenously if there is hypoglycaemia (defined as a blood glucose<3.0 mmol/L). Oxygen should be given by face mask, medical assistance summoned and emergency admission to hospital arranged.
Gonadotropin (GnRH) deficiency
Hyposecretion of GnRH may be seen most commonly in intense physical training or anorexia nervosa, and rarely in syndromes (Box 6.1).
Kallmann syndrome (hypogonadotropin eunuchoidism) is the most frequent form of isolated gonadotropin deficiency – a rare X-linked recessive disease caused by a defect in a cell adhesion protein that participates in the migration of the GnRH neurons. These normally originate in olfactory tissues and migrate to the hypothalamus. Impaired sense of smell is caused by the absence of the olfactory bulbs. Kallmann syndrome mainly affects males and becomes apparent when they fail to develop secondary sexual characteristics. Thus, hypogonadism and anosmia, with underdeveloped genitalia and sterile gonads, are classic presentations. Treatment involves oestrogen or testosterone replacement, and pulsatile GnRH injections.
Anterior Pituitary Hyperfunction
Growth hormone excess: gigantism and acromegaly
General aspects
Overproduction of growth hormone by an anterior pituitary adenoma causes gigantism before the epiphyses have fused, and acromegaly thereafter.
Clinical features
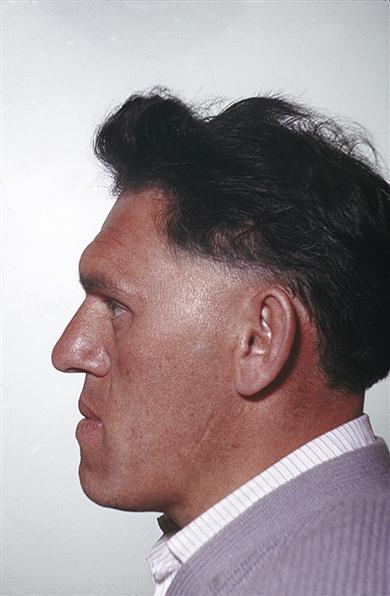
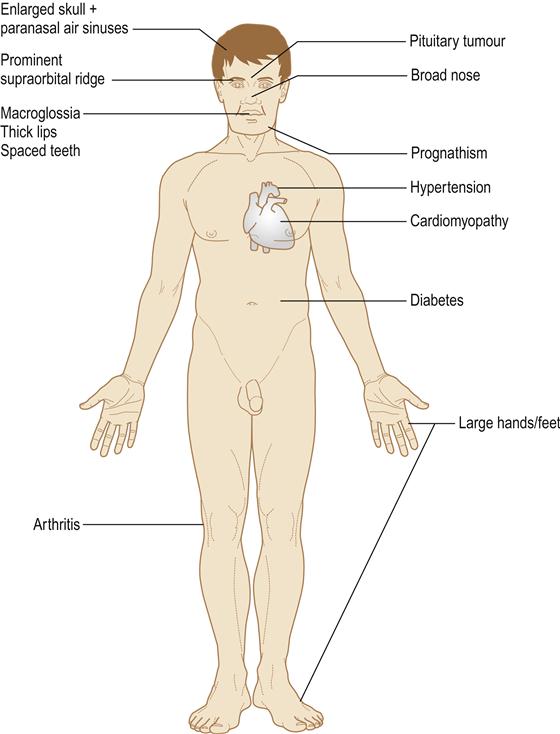
In gigantism, all the organs, soft tissues and skeleton enlarge, leading to excessively tall stature, thickening of the soft tissues with prominence of the supraorbital ridge, coarse oily skin, thick spade-like fingers and deepening of the voice.
In acromegaly, only those bones with growth potential, particularly the mandible, can enlarge. There is thickening of the soft tissues and the hands become large and spade-like. Acromegaly is one of the few endocrine diseases that should be instantly recognized – even in a passer-by – by the gross prognathism and the thickened facial features and hands (Fig. 6.5).
In both gigantism and acromegaly, local pressure effects from the pituitary tumour may cause hypopituitarism plus compression of the optic chiasma, with visual field defects and raised intracranial pressure with severe headaches.
Growth hormone disorders may be complicated by diabetes mellitus, hypertension, cardiomyopathy, sleep apnoea, hypercalcaemia and osteoarthritis (Fig. 6.6). Life expectancy is also shortened by diseases such as colonic carcinoma.
General management
Family photographs may clearly demonstrate the increasingly coarse features. Skull radiography, computed tomography (CT) and MRI scans (for pituitary – sella turcica – enlargement) are indicated. Sella enlargement is also sometimes due to other causes, such as hypothalamic masses or cysts or the empty sella syndrome (this is caused by herniation of a sac of leptomeninges into the sella). Rhinorrhoea may warrant surgical correction, but no other treatment is needed.
Other investigations in growth hormone excess are visual field assessment (to detect optic chiasm involvement), glucose tolerance tests (to exclude diabetes and to assess the plasma growth hormone response), levels of growth hormone and insulin-like growth factor 1 (IGF-1), and assessment of function of the remaining pituitary.
The pituitary adenoma may be resected trans-sphenoidally or the whole gland may have to be irradiated (e.g. by transnasal yttrium). Hypopituitarism or diabetes insipidus follows such treatment.
Bromocriptine or octreotide (an analogue of the hypothalamic release-inhibiting hormone somatostatin) may also help treatment.
Dental aspects
Local analgesia is suitable. CS may be given, if necessary, provided the airway is clear. GA may be hazardous because of complications (see later).
Dental management may be complicated by:
Mandibular enlargement leads to prognathism (class III malocclusion) with spacing of the teeth and thickening of all soft tissues, but most conspicuously of the face. Orthognathic surgery may be needed and fatalities have followed such surgery in the past because of airway obstruction. The paranasal air sinuses are enlarged and the skull thickened. Sialosis may develop.
Pancreas
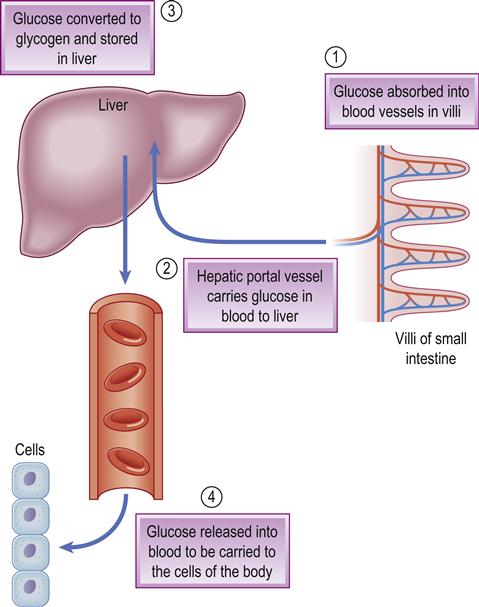
All carbohydrate eaten is digested into glucose, which passes from the stomach and intestine into the blood and is the main source of energy. Glucose homeostasis depends mainly on the pancreatic hormones insulin (from the beta-cells of the islets of Langerhans) and glucagon from the alpha-cells in the islets.
Insulin facilitates glucose transport into cells and thus lowers blood sugar (glucose) levels. Glucagon stimulates the liver to convert glycogen into glucose, thus raising glucose levels and having the opposite effect to insulin (Fig. 6.7). The beta-cells also produce amylin, which modulates gastric emptying and satiety, and decreases the postprandial rise in glucagon. Also involved in glucose homeostasis are gastric inhibitory polypeptide (GIP) and glucagon-like peptide 1 (GLP-1) – small intestine hormones (incretins) secreted when eating. GLP-1 has a glucose-lowering effect via stimulation of insulin production and inhibition of glucagon (as well as preserving pancreatic beta-cell function, slowing gastric emptying and suppressing food intake). Several other hormones that can influence glucose include catecholamines (adrenaline [epinephrine] and noradrenaline [norepinephrine]), corticosteroids, thyroid hormone and growth hormone.
Diabetes Mellitus
General aspects
Diabetes mellitus (DM) is a disorder caused by an absolute or relative lack of insulin; there can be a low output of insulin from the pancreas, or the peripheral tissues may resist insulin. In diabetes, with insulin lacking or its action blocked, glucose cannot enter cells and, without energy, weakness results. Glucose also then accumulates in the blood (hyperglycaemia) and spills over into the urine (glucosuria), taking with it, osmotically, a large amount of water (polyuria). This leads to dehydration and thus thirst and the need to drink excessively (polydipsia). As glucose is then no longer a viable energy source, fat and protein stores are metabolized with weight loss, peripheral muscle wasting and, in type 1 diabetes, the production of ketone bodies (acetoacetate, β-hydroxybutyrate and acetone). In severe cases, ketone bodies may be detected on the breath (in particular, acetone) and accumulate in the blood (ketonaemia), as well as being excreted in the urine (ketonuria). The resultant metabolic ketoacidosis leads to a compensatory increase in respiratory rate (hyperventilation) and a secondary respiratory alkalosis.
Chronic hyperglycaemia causes microvascular complications and atherosclerosis, and is a leading cause of death and disability.
Diabetes affects about 3–4% of the general population but may be recognized in only 75% of those individuals, yet it is a leading cause of death and disability. Worldwide, about 246 million people are affected. Risk factors for diabetes include family history (the risk of developing diabetes rises if a close relative, such as a parent or sibling, has the disease); being overweight; inactivity; age (the risk of developing type 2 diabetes rises with age, especially after age 45); and race. Genetics has a role. Type 1 diabetes is more common in Caucasians and in European countries, such as Finland and Sweden. Type 2 diabetes is especially common in people of African heritage, Asians and Hispanics, and has a stronger genetic background. Diabetes affects up to one-third of the elderly from the Asian subcontinent. Among the Pima Indians in the USA and in Nauru in the Pacific, around half of all adults have type 2 diabetes – the highest rates in the world. The prevalence of diabetes appears to be rising, mainly as more children and adolescents become overweight. In contrast, fewer diabetics remain undiagnosed, and associated ischaemic heart disease and retinal disease are falling.
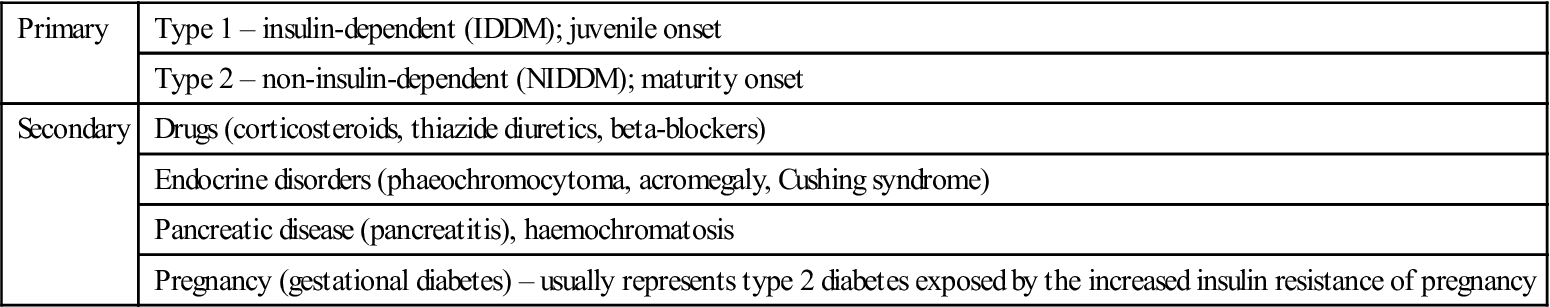
Diabetes may be primary, or secondary to some other factor(s), as shown in Table 6.5. Type 2 is by far the most common type (Table 6.6).
Table 6.5
Classification of diabetes mellitus
< ?comst?>
| Primary | Type 1 – insulin-dependent (IDDM); juvenile onset |
| Type 2 – non-insulin-dependent (NIDDM); maturity onset | |
| Secondary | Drugs (corticosteroids, thiazide diuretics, beta-blockers) |
| Endocrine disorders (phaeochromocytoma, acromegaly, Cushing syndrome) | |
| Pancreatic disease (pancreatitis), haemochromatosis | |
| Pregnancy (gestational diabetes) – usually represents type 2 diabetes exposed by the increased insulin resistance of pregnancy |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
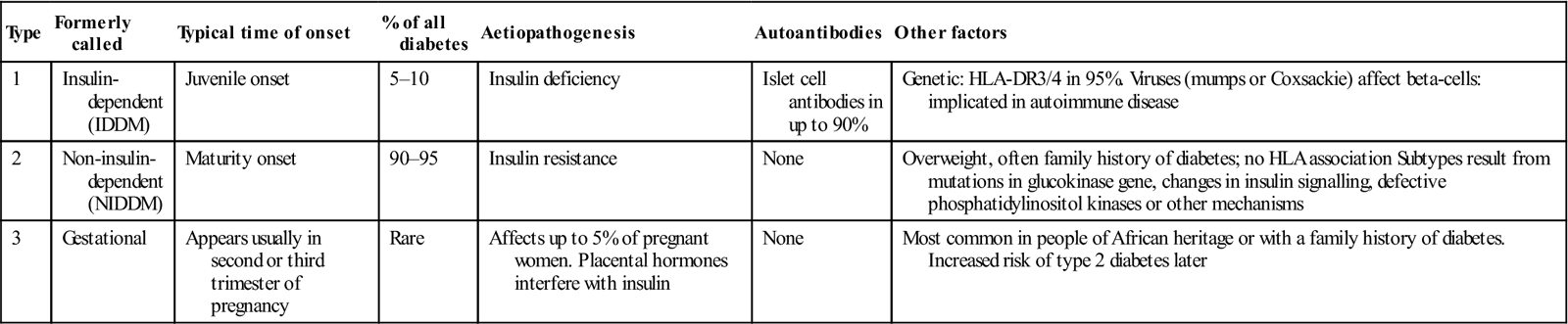
Table 6.6
< ?comst?>
| Type | Formerly called | Typical time of onset | % of all diabetes | Aetiopathogenesis | Autoantibodies | Other factors |
| 1 | Insulin-dependent (IDDM) | Juvenile onset | 5–10 | Insulin deficiency | Islet cell antibodies in up to 90% | Genetic: HLA-DR3/4 in 95%. Viruses (mumps or Coxsackie) affect beta-cells: implicated in autoimmune disease |
| 2 | Non-insulin- dependent (NIDDM) | Maturity onset | 90–95 | Insulin resistance | None | Overweight, often family history of diabetes; no HLA association Subtypes result from mutations in glucokinase gene, changes in insulin signalling, defective phosphatidylinositol kinases or other mechanisms |
| 3 | Gestational | Appears usually in second or third trimester of pregnancy | Rare | Affects up to 5% of pregnant women. Placental hormones interfere with insulin | None | Most common in people of African heritage or with a family history of diabetes. Increased risk of type 2 diabetes later |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Type 1 diabetes (T1DM)
Type 1 diabetes develops when the immune system attacks insulin-producing cells of the pancreatic islets of Langerhans. Weight reduction and exercise increase sensitivity to insulin, thus helping to control blood glucose elevations, but these patients still require insulin.
Type 1 diabetes – formerly termed insulin-dependent (IDDM) or juvenile-onset diabetes – is most commonly diagnosed at about 12 years of age and commonly presents before the third decade, but can appear at any age. Associated with other organ-specific autoimmune diseases, it is characterized by antibodies directed against insulin and the pancreatic islets of Langerhans. It may have a viral (possibly Coxsackie or rubella) aetiology. Latent autoimmune diabetes in adults (LADA) is essentially a slow presentation of type 1 diabetes, which is seen in slimmer patients who progress quickly to insulin requirement. Anti-glutamic acid decarboxylase (GAD) antibodies may be helpful in the diagnosis.
Type 2 diabetes (T2DM)
Type 2 diabetes – formerly termed non-insulin-dependent (NIDDM) or maturity-onset diabetes – accounts for at least 90% of diabetics. Generally, it occurs in genetically predisposed individuals over the age of 40, who are typically overweight. Patients are insulin-resistant and have diminished beta-cell function. Alcohol binge drinking may induce insulin resistance independent of caloric intake and increase the risk for developing metabolic syndrome and T2DM. Maturity-onset diabetes of the young (MODY) is a rare form of type 2 diabetes; it caused by autosomal dominant mutations and so there is vertical transmission of diabetes within families. The phenotype varies from mild glucose intolerance to insulin-requiring diabetes, typically diagnosed before the age of 25 years.
Type 3 diabetes (T3DM)
Gestational diabetes is basically an insulin-resistant state exposing the patient to a future risk of type 2 diabetes (see Table 6.6).
Clinical features
Patients with diabetes may be asymptomatic and detected on routine or opportunistic screening, or present in a variety of ways related to severity and degree of onset (Table 6.7).
Table 6.7
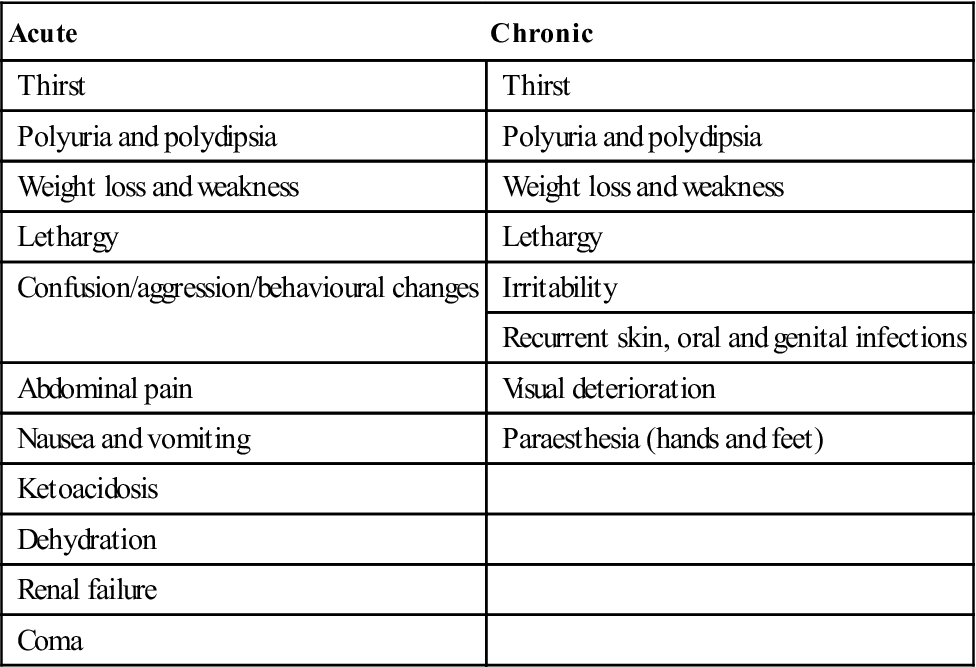
Acute and chronic presenting features of diabetes mellitus
< ?comst?>
| Acute | Chronic |
| Thirst | Thirst |
| Polyuria and polydipsia | Polyuria and polydipsia |
| Weight loss and weakness | Weight loss and weakness |
| Lethargy | Lethargy |
| Confusion/aggression/behavioural changes | Irritability |
| Recurrent skin, oral and genital infections | |
| Abdominal pain | Visual deterioration |
| Nausea and vomiting | Paraesthesia (hands and feet) |
| Ketoacidosis | |
| Dehydration | |
| Renal failure | |
| Coma |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Lethargy is the most common symptom but hyperglycaemia, polyuria and thirst (polydipsia) are prominent. Since glucose is lost as an energy source in type 1 diabetes, fats must be metabolized, leading to weight loss from fat breakdown to fatty acids and ketone bodies, which appear in the blood, causing acidosis and hyperventilation, in the urine (ketonuria) and to some extent in the breath (acetone). Overproduction of acetoacetate is converted to the other ketone bodies, hydroxybutyrate and acetone. Diabetes is also associated with immune deficiencies, particularly polymorph dysfunction, leading to susceptibility to infections (mainly skin infections and mucosal candidosis; Fig. 6.8).
Type 1 diabetes develops most often in children and young adults, generally before the age of 25. Symptoms usually develop over a short period. Excessive thirst and urination, constant hunger, weight loss, blurred vision and extreme fatigue are typical. Life-threatening diabetic coma (diabetic ketoacidosis) is a significant risk. Insulin is required daily for treatment, for life, and diet must be controlled. Hypoglycaemia is a risk.
Type 2 diabetes develops most often in overweight patients. Symptoms usually develop gradually. Fatigue, frequent urination, unusual thirst, weight loss, blurred vision, frequent infections, and slow healing of wounds or sores are seen. Most patients with type 2 diabetes can be managed on diet and oral hypoglycaemic drugs. Many will eventually need insulin but are often resistant. About 80% of people with type 2 diabetes have the metabolic syndrome, which includes obesity, elevated blood pressure and high levels of blood lipids (Ch. 23).
Factors that affect the blood glucose level are shown in Box 6.2.
Acute complications
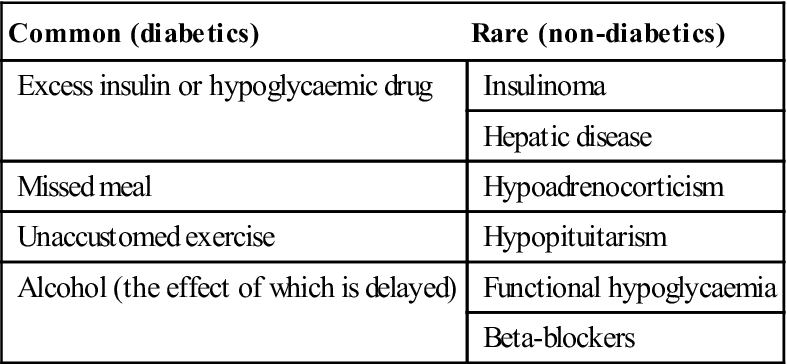
Diabetes can lead to coma. Hypoglycaemic coma is the main acute complication of diabetes, is growing in frequency with the trend towards tighter metabolic control of diabetes, and is usually the result of one or more of the above factors. Less common causes are shown in Table 6.8. Many insulin-treated patients are liable to hypoglycaemia, due to an imbalance between food intake and usage, and insulin therapy. Hypoglycaemia can be of rapid onset and may resemble fainting. Adrenaline (epinephrine) is released, leading to a strong and bounding pulse, sweaty skin, and often anxiety, irritability and disorientation, before consciousness is lost. Occasionally, the patient may convulse.
Table 6.8
< ?comst?>
| Common (diabetics) | Rare (non-diabetics) |
| Excess insulin or hypoglycaemic drug | Insulinoma |
| Hepatic disease | |
| Missed meal | Hypoadrenocorticism |
| Unaccustomed exercise | Hypopituitarism |
| Alcohol (the effect of which is delayed) | Functional hypoglycaemia |
| Beta-blockers |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
The patient should be treated immediately (Table 6.9); hypoglycaemia must be quickly corrected with glucose or brain damage can result. Glucose will cause little harm in hyperglycaemic coma but will improve hypoglycaemia. Never give insulin since this can cause severe brain damage or kill a hypoglycaemic patient. Assess the glucose level with a testing strip.
Table 6.9
Hypoglycaemic and hyperglycaemic comas and their treatment
< ?comst?>
| Hypoglycaemic coma | Hyperglycaemic coma |
| Diabetes usually known | Diabetes may be unrecognized |
| Too much insulin or too little food, or too much exercise or alcohol | Too little insulin, infection or myocardial infarct |
| Management | |
| Take blood for baseline glucose, electrolytes, urea, Hb and packed cell volume (PCV) | |
| If conscious, give Hypostop (GlucoGel) or 25 g glucose orally (2 teaspoons sugar, 3 lumps sugar, 3 Dextrosol tablets, 60 mL Lucozade, 15 mL Ribena, 90 mL cola drink (not ‘diet’ variety) or 170 mL milk) | Put up infusion to rehydrate and give insulin |
| If comatose, give 20 mg 20% or 50% dextrose i.v. and on arousal 25 mg orally | |
| If i.v. injection impossible, give glucagon 1 mg i.m. | |
| Call ambulance (Ch. 1) | |
< ?comen?>< ?comst1?>

< ?comst1?>
< ?comen1?>
If the patient is conscious, give glucose solution or gel (GlucoGel) immediately by mouth or 10 g sugar but, if the patient is comatose, give 10–20 mL of 20–50% sterile dextrose intravenously or, if a vein cannot readily be found, glucagon 1 mg intramuscularly. On arousal, the patient should also be given glucose orally, usually in the form of longer-acting carbohydrate (e.g. bread, biscuits).
Other diabetics are difficult to control (brittle diabetes – a term usually used in association with adolescent girls who may be self-harming) and more prone to ketosis, severe acidosis and hyperglycaemia (diabetic coma), the result of a relative or absolute deficiency of insulin. In patients under treatment, it may be precipitated by factors such as infections.
Hyperglycaemic coma usually has a slow onset over many hours, with deepening drowsiness (but unconsciousness is rare, so an unconscious diabetic should always be assumed to be hypoglycaemic), signs of dehydration (dry skin, weak pulse, hypotension), acidosis (deep breathing), and ketosis (acetone smell on breath and vomiting) mainly in type 1 diabetes. If it is certain that collapse is due to hyperglycaemic ketoacidotic coma, the immediate priority is to establish an intravenous infusion line. This enables rapid rehydration to correct dehydration and electrolyte (especially potassium) losses, and the administration of insulin. Blood should be taken for baseline measurements of glucose, electrolytes, pH and blood gases. Raised plasma ketone body levels can be demonstrated with a testing strip such as Ketostix (Ames). Insulin is then started, such as 20 units i.m. stat. Medical help should be obtained as soon as possible.
Coma in a diabetic patient, though usually due to hypo- or hyperglycaemia, may have other causes such as myocardial infarction (Ch. 5).
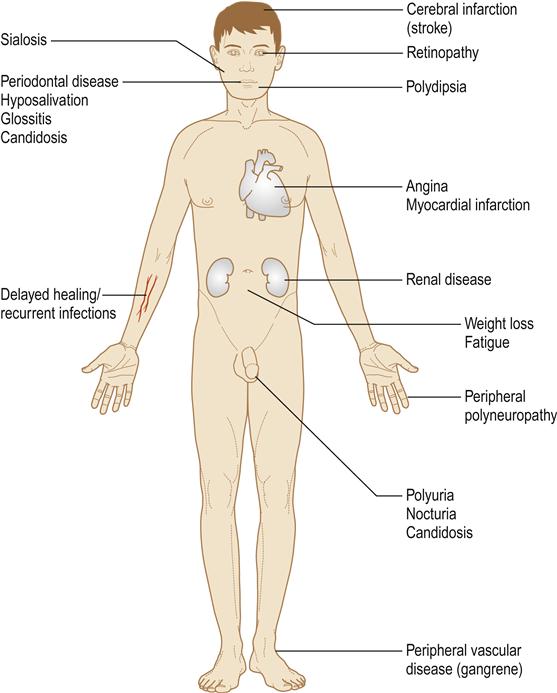
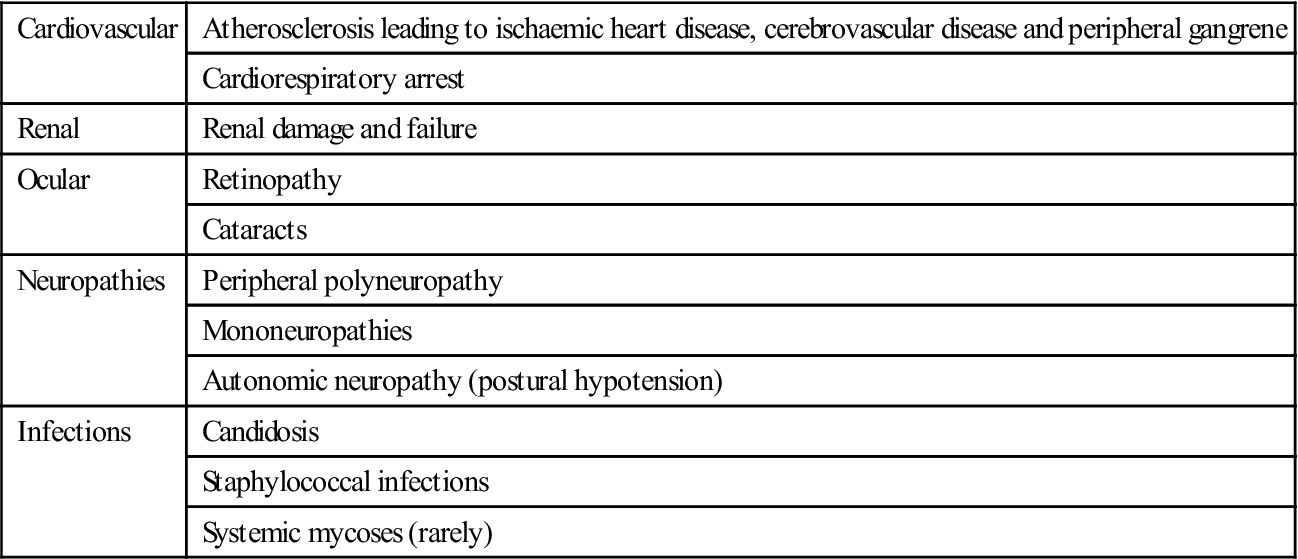
Chronic complications
Diabetes is associated with long-term microvascular and macrovascular complications that can affect almost every part of the body (Table 6.10). It is a leading cause of death and disability due to premature cardiovascular disease; the risk of myocardial infarction is at least tripled. Renal and retinal complications are also incapacitating, as is gangrene of toes.
Table 6.10
Chronic complications of diabetes
< ?comst?>
| Cardiovascular | Atherosclerosis leading to ischaemic heart disease, cerebrovascular disease and peripheral gangrene |
| Cardiorespiratory arrest | |
| Renal | Renal damage and failure |
| Ocular | Retinopathy |
| Cataracts | |
| Neuropathies | Peripheral polyneuropathy |
| Mononeuropathies | |
| Autonomic neuropathy (postural hypotension) | |
| Infections | Candidosis |
| Staphylococcal infections | |
| Systemic mycoses (rarely) |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Smoking increases the risk of many chronic complications. Phagocytic defects contribute to an immune defect and liability to infections. Babies born to women with diabetes are often large and sometimes have birth defects. Occasionally, there are associations of type 1 diabetes with autoimmune disorders, especially Addison disease. There may be a predilection to oral cancer.
General management
Glycosuria is usually indicative of diabetes, but absence of glycosuria does not completely exclude it and confirmation by blood glucose levels is essential. Diagnosis is from the presence of raised blood (venous plasma) glucose level (Table 6.11).
Table 6.11
Glucose assays in the diagnosis of diabetes
| Test | Confirms diabetesa | Excludes diabetes |
| Fasting plasma glucose (after a person has fasted for 8 h) | >7.0 mmol/L | <6 mmol/L |
| Random blood glucose (taken at any time of day) | ≥11.1 mmol/L | <8 mmol/Lb |
| Plasma glucose taken 2 h after a person has consumed a drink containing 75 g of glucose in oral glucose tolerance test (OGTT) | >11.1 mmol/La | <11.1 mmol/L |
aIf associated with symptoms, or abnormal on repeat testing.
A diagnosis of diabetes is made when any one of the glycosuria, random blood glucose and fasting blood glucose tests is positive, with a second test positive on a different day (not needed if symptomatic). Pre-diabetes is the term applied when there is either of the following:
Patients with IGT and IFG are more likely to develop type 2 diabetes, and have an increased risk of cardiovascular disease.
Prevention
■ Weight should be reduced and physical activity stepped up.
■ Diet may help diabetic control; recommendations are to:
Eat more starches, such as bread, cereal and starchy vegetables
Treatment
■ reduction of hyperglycaemia – to alleviate presenting symptoms
■ prevention of hypoglycaemia and other acute complications
■ maintenance of near-normal blood glucose levels – to minimize microvascular complications
■ reduction of other cardiovascular risk factors (smoking, exercise, hypertension and lipids).
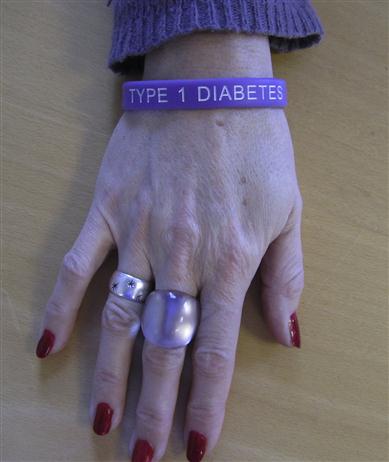
Individuals should be educated about the importance of good long-term diabetic control; they should have a thorough knowledge of the disease and may wish to warn others that they have it (Fig. 6.9). All diabetics should carry a card indicating the diagnosis, treatment schedule and physician in charge, and should be encouraged to wear a MedicAlert-type device.
The progressive nature of diabetes, its complications and management aims should be well understood. Patients should also be aware of the range of diabetic services available and how to make appropriate use of them. A multidisciplinary team approach is required for their effective management. Meals should be taken at regular intervals, with a high fibre and relatively high carbohydrate content but avoiding sugars, and with a caloric intake strictly related to physical activity. Lifestyle changes, such as exercise, diet, fasting, travel (including changing time zones) and changes in working practices, can have an impact on diabetic control.
Urine testing has little or no place in the assessment or control of diabetes. Control is assessed by blood glucose levels and used to guide medication. A glucometer allows glucose readings to be taken easily by the patient at home. Fasting glucose<6 mmol/L is normal. Long-term assessment of glucose control is made by estimation of the blood level of glycosylated haemoglobin (HbA1c). HbA1c is normal adult haemoglobin that binds glucose and remains in the circulation for the life of the erythrocyte, therefore acting as a cumulative index of diabetic control over the preceding 3 months; the higher the glycosylated haemoglobin level, the greater the risk of chronic complications. Patients should have an HbA1c test every 3–6 months; glycosylated haemoglobin below 4.8% of total haemoglobin is normal. Fructosamine is an alternative assay of long-term diabetic control.
Optimum control would be to aim to keep blood glucose levels at 4–7 mmol/L before meals (preprandial) and at no higher than 10 mmol/L 2 hours after meals (postprandial), and HbA1c (long-term glucose level) at 7% or less.
Type 1 diabetes
Basic treatment of type 1 diabetes is subcutaneous insulin, the amount being balanced against food intake and daily activities, and monitored via home blood glucose testing. Insulin cannot be taken orally but must be given via injection, subcutaneously, either in the abdominal wall (Fig. 6.10), buttocks, thighs or upper arms, using a syringe, pen device or insulin pump. An artificial pancreas, a “closed loop” system that monitors blood glucose levels to adjust the insulin being administered by a pump is being developed. There are three groups of insulins – animal, human (actually synthetic) and analogues. There are six main types of insulin, available in various combinations and all working in different ways (Table 6.12). Most insulin regimes are based on a long-acting insulin combined with a smaller amount of faster-acting insulin, all normally taken via two or more injections each day. Insulin pump therapy (continuous subcutaneous insulin infusion [CSII]) is fast-acting insulin given continually day and night, at a rate preset according to needs. An insulin pump is not a closed loop system; rather, patients learn to set the dose according to diet, activity and blood glucose levels. Most test a minimum of four times a day, in order to obtain the information needed to set an appropriate insulin dose. An advantage of insulin pump therapy is that it can help improve control and minimize the frequency of ‘hypos’. The types of insulin used include those shown in Table 6.12.
Table 6.12
< ?comst?>
| Insulin type | Protocol | Peak activity | Approximate duration of activity |
| Rapid-acting analogues | Injected just before, with or after food | 0–3 h | 2–5 hours |
| Long-acting analogues | Injected once a day, not necessarily with food | – | 24 h |
| Short-acting insulins | Injected 15–30 min before a meal to cover the rise in blood glucose levels that occurs after eating | 2–6 h | 8 h |
| Medium- and long-acting insulins | Injected once or twice a day to provide background insulin or in combination with short-acting insulins/rapid-acting analogues | 4–12 h | 30 h |
| Mixed insulin | Combination of medium- and short-acting insulin | Varies | Varies |
| Mixed analogue | Medium-acting insulin and rapid-acting analogue | Varies | Varies |
< ?comen?>< ?comst1?>

< ?comst1?>
< ?comen1?>
Type 2 diabetes
Medications for type 2 diabetes are designed to:
■ increase the pancreatic output of insulin
■ increase the response of cells to insulin
When selecting therapy, consideration should be given to:
■ the magnitude of change in blood glucose control
Some people with type 2 diabetes find that, despite having their diabetes medication adjusted, their blood glucose levels remain too high />
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


