Fig. 10.1
Humans are far more different from each other in their microbial composition than in their genomic composition. The colors on the left side of each individual represent bacterial phyla, while the colors on the right side indicate host genomic similarity. For the most part we contain similar phyla living in and on our bodies, including the oral cavity, but their relative abundance can be drastically different. On the other hand, our genomic composition is nearly identical, with only a small fraction (ca 0.1 %) differing across individuals (Adapted from Califf et al. 2014)
10.1.1 Bacteria
Bacteria have been considered the dominating part of the microbiome in man. However, while some six billion bacteria are present in the oral cavity, it contains potentially 35 times that many bacteriophages/viruses (Edlund et al. 2015). When Dewhirst et al. (2010) established the Human Oral Microbiome Database (HOMD) (http://www.homd.org/), it comprised over 600 prevalent bacterial taxa at the species level with distinct subsets predominating at different sites such as teeth, gingival sulcus, tongue, cheeks, hard/soft palate, and tonsils. The HOMD included 619 taxa from 13 phyla: Actinobacteria, Bacteroidetes, Chlamydiae, Chloroflexi, Euryarchaeota, Firmicutes, Fusobacteria, Proteobacteria, Spirochaetes, SR1, Synergistetes, Tenericutes, and TM7. The analysis comprised 1179 taxa. Among these 24 % were named, 8 % were cultivated but unnamed, and 68 % were uncultivated phylotypes. Later the number of oral phyla has been extended to 15, but 96 % of the sequences are accounted for by only 6 phyla: Actinobacteria, Bacteroidetes, Firmicutes, Fusobacteria, Proteobacteria, and Spirochaetes (Wade 2013). Recently, Camanocha and Dewhirst (2014) developed primer pairs for making phylum-selective 16S rRNA clone libraries and identified species from the lesser known oral phyla or candidate divisions including Synergistetes, TM7, Chlorobi, Chloroflexi, GN02, SR1, and WPS-2.
10.1.2 Bacteriophages/Viruses
Oral viruses in saliva are dominated by bacteriophages (Pride et al. 2012). Also dental plaque is inhabited by a community of bacteriophages (Naidu et al. 2014). Bacteriophages constitute the major part of the oral virome with relatively few eukaryotic viruses identified such as herpesviruses, papillomaviruses, enteroviruses, and circoviruses (Grinde and Olsen 2010; Naidu et al. 2014). The mouth has been found to have more genetic elements than the stool, i.e., viruses, plasmids, and transposons, although it has fewer bacteria (Zhang et al. 2013). Bacteriophages may serve as reservoirs for genes functioning in the oral cavity. Phage members of the oral virome can carry genes involved in resistance to complement degradation of immunoglobulins, adhesion to cells lining the oropharynx, and antibiotic resistance (Pride et al. 2012; Muniesa et al. 2013; Abeles et al. 2014; Quirós et al. 2014).
Oral viruses have gene functions that may be involved in the pathogenic roles of their host bacteria (Pride et al. 2012). The same salivary viruses could be identified at all time points over 60 days despite being present in low numbers (Abeles et al. 2014), reflecting that the oral viral ecosystem is stable. Most oral viruses are lysogenic and live in harmony with their hosts (Abeles and Pride 2014; Ly et al. 2014), and they may be important in shaping the microbial diversity of the oral cavity. Another peculiarity is that viral communities of the mouth are highly personalized (Willner et al. 2011; Pride et al. 2012), even more personalized than bacterial communities when analyzed with 16S rDNA sequencing (Abeles et al. 2014). A noteworthy feature is also that oral viruses vary according to host sex, rather than among individuals (Abeles et al. 2014). The human oral viral community is probably a result of the unique viral exposures of each individual (Abeles et al. 2014), but considerably more of the oral virobiota of people living together is shared than could be expected by chance (Robles-Sikisaka et al. 2013). Eukaryotic viruses such as Torque Teno viruses (TTVs) and SEN viruses have been found in the bloodstream of healthy people (Pride et al. 2012; Abeles and Pride 2014). Blood of healthy persons have previously been considered sterile. Both these groups of viruses are present in the human oral cavity (Pride et al. 2012). Also herpesviruses, shed in the mouth from healthy individuals, can be found in human blood (De Vlaminck et al. 2013). Therefore not only bacteria but also viruses can translocate through mucosal surfaces to the bloodstream and possibly be involved in systemic diseases.
It is well known that the human oral cavity contains a large and diverse variety of bacteria. What viruses it contains has to a great extent been overlooked. This particularly relates to the periodontal microbiota, although herpesviruses including Epstein-Barr virus and cytomegalovirus can be present in high copy counts in aggressive periodontitis and may interact with periodontopathogenic bacteria to cause the disease (Sunde et al. 2008; Slots 2011; Contreras et al. 2014). Ly et al. (2014) examined samples from saliva of periodontally healthy and diseased patients and found that the communities of viruses inhabiting saliva and subgingival and supragingival biofilms were composed mainly of bacteriophages. The virome composition was greatly reflected by the site it was collected from. The largest difference in composition was between supra-/subgingival plaque and saliva. Differences in virus composition were significantly related to the health status of viruses in plaque, but not to those in saliva. Noteworthy, there was a significant increase in myoviruses (generally lytic) in subgingival biofilm suggesting that these viruses may have a great importance to local bacterial diversity and that the virus may serve as useful indicators of the oral health status. Since viruses have the potential to form microbial communities as well as to elicit host immune response, they probably play an important role in human health (Edlund et al. 2015). Also, the fact that they are personal, persistent, and gender specific suggests that they can be important in the interplay between host genetics and the environment.
10.1.3 Archaea
Archaea were originally considered a primitive form of life that thrives in extreme environments. However, high numbers of methane-producing archaea (methanogens) have now been detected in the oral cavity (Belay et al. 1988), the gastrointestinal tract (Karlin et al. 1982), and vagina (Belay et al. 1990) of human beings. The reported oral archaea contain the genera Methanobrevibacter, Methanobacterium, Methanosarcina, and Methanosphaera and the order Thermoplasmatales (He et al. 2014). The main species is Methanobrevibacter oralis. Archaea have been detected in saliva, periodontitis, peri-implantitis, pericoronitis, and infected root canals (Brusa et al. 1987; Belay et al. 1988; Kulik et al. 2001; Lepp et al. 2004; Vianna et al. 2006, 2009; Vickerman et al. 2007; Conway de Macario and Macario 2009; Jiang et al. 2009; Matarazzo et al. 2011, 2012; Faveri et al. 2011; Mansfield et al. 2012; Bringuier et al. 2013). These studies detected a higher frequency of archaea in oral infections than in health. Thus the relative abundance of archaea in subgingival plaque increased with the severity of periodontitis and decreased with the reduction of periodontitis after treatment. Archaea may therefore be associated with periodontitis but the diversity of archaea is limited (Li et al. 2009). Almost all sequenced amplicons fell in the genus Methanobrevibacter of the Euryarchaeota phylum with M. oralis-like species as the most dominant. In root canal infections, presence of archaea was associated with clinical symptoms (Jiang et al. 2009). Although discussion of the clinical role of Euryarchaeota (including Methanobrevibacter smithii, M. oralis, and Methanosphaera stadtmanae) continues (Horz and Conrads 2010), and archaea are emerging organisms in complex human microbiomes (Dridi et al. 2011), methanogenic archaea do not seem to induce oral diseases directly. However, they may promote anaerobic infections through syntropic interactions with true pathogenic fermenting bacteria, e.g., through interspecies H2 transfer, thereby favoring growth of certain bacteria (Matarazzo et al. 2012). Thus, a positive correlation has been found between methanogens and Synergistes species in oral infections (Vianna et al. 2006; Vartoukian et al. 2007).
10.1.4 Fungi
Dupuy et al. (2014) performed massive parallel, high-throughput sequencing of internal transcribed spacer 1 (ITS1) amplicons from saliva after robust extraction methods. Their findings confirmed nearly every community member from a similar study by Ghannoum et al. (2010) who had detected 74 cultivable and 11 non-cultivable fungal genera in the oral cavity by using multitag pyrosequencing of panfungal ITS primers. A consensus on genus-level members of oral fungi (core mycobiome) was thereby reached. This study was the first to demonstrate not-yet-cultivated fungi in the oral cavity. It was suggested that such organisms could be the reason for failure in the treatment of oral fungal infections. Consensus members of the saliva microbiome were Candida/Pichia, Cladosporium/Davidiella, Alternaria/Lewia, Aspergillus/Emericella/Eurotium, Fusarium/Gibberella, Cryptococcus/Filobasidiella, and Aureobasidium. Weaker candidates for consensus inclusion were Saccharomyces, Epicoccum, and Phoma. Interestingly, Malassezia species, that are important commensals of human skin, were for the first time included in the oral core mycobiome. The oral fungal community showed a consistent intraindividual stability over time, but there was high interindividual variability (Monteira-da-Silva et al. 2014).
Interactions between fungi and bacteria, e.g., between Candida and streptococci, may influence oral health (Diaz et al. 2014). A symbiotic relationship between S. mutans and C. albicans has been found to synergize virulence of plaque biofilms in vivo (Falsetta et al. 2014). Thus S. gordonii glucosyltransferase promotes biofilm interactions with C. albicans (Ricker et al. 2014). Fungi probably have a role in maintaining a balance between microorganisms and the host (Krom et al. 2014).
10.1.5 Protozoa
Protozoa are parts of the normal microbiome. The best known are Entamoeba gingivalis and Trichomonas tenax (Vozza et al. 2005). They are present in subjects who neglect their oral hygiene and predominantly in subgingival plaque from patients with periodontal disease (Lange et al. 1983). Both have been linked to gingivitis and they were once considered pathogens. T. tenax has been correlated with xerostomia, burning mouth, and periodontal pockets (Kurnatowska 1993; Kurnatowska and Kurnatowski 1998). Later, it has become clear that these organisms increase when the oral hygiene deteriorates. Their increase may be due to nutrients accessible from debris and bacteria (Wade 2013). It is interesting though that metronidazole, frequently used as an effective supplement in the treatment of periodontitis, is active against both Entamoeba and Trichomonas.
10.2 Techniques to Analyze the Oral Microbiota
It should be realized that every technique that has been used to detect oral microorganisms has its strengths and limitations. Not all of these techniques will be dealt with here. Microscopy and culture were long standard methods for assessment of the oral microbiota. Later, culture helped us become more familiar with this microbiota when methods for recovery of anaerobic bacteria were developed. However, it soon became clear that only half of the oral microbiota could be cultured. Therefore culture-independent methods were exploited, particularly DNA-DNA hybridization and PCR-based assays. DNA-DNA hybridization (checkerboard) relied though on bacteria that could be cultivated for the making of whole genomic probes (Socransky et al. 1994), but reverse-capture checkerboard hybridization did not (Paster et al. 1998). Checkerboard DNA-DNA hybridization was helpful delineating bacteria clinically related to periodontitis such as the red and the orange complex (Socransky et al. 1998). Since there was reason to believe that also not-yet-cultivated bacteria could be involved in disease methods, targeting the small subunit (16S) ribosomal RNA molecule was used. These efforts have provided a vast amount of knowledge and description of the oral microbiota. They have also shown that the oral microbiota is not uniform but varies from site to site (Fig. 10.2). The information has been collected in the first curated collection of a human-associated microbiome, HOMD, which provides a description of the organisms and their genomics together with a 16S rRNA identification tool (Dewhirst et al. 2010), and later in the CORE database that is a phylogenetically curated 16S rDNA database of the core oral microbiome (Griffen et al. 2011). Although 16S rRNA gene amplification and Sanger sequencing significantly increased our knowledge of the major components of the oral microbiota, they did not provide information of the entire microbiota. Organisms that are present in low amounts were first revealed by pyrosequencing (next-generation sequencing methods).
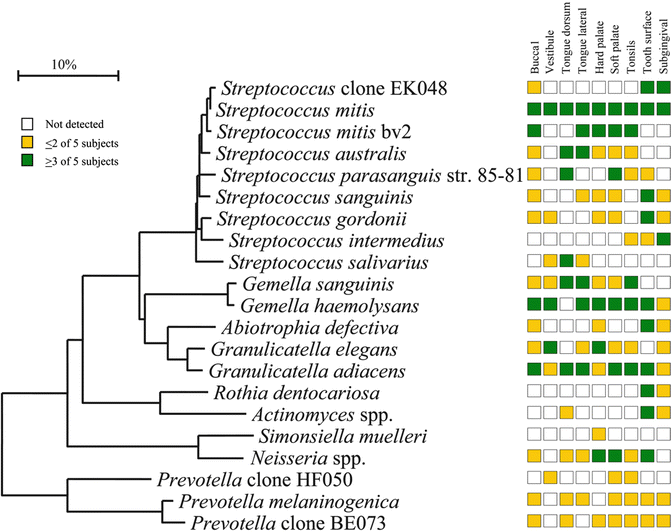
Fig. 10.2
Site specificity of predominant bacterial species in the mouth. Bacterial species or phylotypes were selected on the basis of their detection in multiple subjects for a given site. Distributions of bacterial species in oral sites among subjects are indicated by the columns of boxes to the right of the tree as follows: not detected in any subject (clear box), < 15 % of the total number of clones assayed (yellow box), and ≥ 15 % of the total number of clones assayed (green box). The 15 % cutoff for low and high abundance was chosen arbitrarily. Marker bar represents a 10 % difference in nucleotide sequences (From Aas et al. 2005)
10.2.1 HOMINGS
HOMINGS (http://homings.forsyth.org) apply the speed and efficiency of the next-generation sequencing using the Illumina platform. Almost 600 oral bacterial taxa can be identified with this technique which provides genus-level identification of the remaining sequences for 129 genera. It is thus more comprehensive than its predecessor HOMIM which gave simultaneous microarray detection of about 270 of the most prevalent, cultivated, and not-yet-cultivated oral bacterial species.
10.2.2 Oligotyping Analysis of the Human Oral Microbiome
A limited taxonomic resolution has often prevented understanding the census of bacterial populations in healthy individuals. By using 16S rRNA gene sequence data from nine sites in the oral cavity, Eren et al. (2014) identified 493 oligotypes from their V1-V3 data and 360 oligotypes from the V3-V5 data. The oligotypes were associated with species-level taxon names by comparing with HOMD. The authors discovered closely related oligotypes differing sometimes by only a single nucleotide that showed widely different distributions among oral sites and samples. Different habitat distributions of closely related oligotypes indicated a level of ecological and functional biodiversity not recognized previously. This technique combined with Shannon entropy has the capacity to analyze entire microbiomes and discriminate between closely related but distinct taxa in different habitats.
10.2.3 High-Throughput Sequencing (Pyrosequencing)
16S rRNA sequencing using next-generation sequencing has provided a wealth of new knowledge on the genetic composition of the oral microbiome in health and disease. The most useful of these approaches have relied on the 454 (Roche) pyrosequencing platform. In Table 10.1, the advantages and limitations of different high-throughput sequencing platforms are summarized.
Table 10.1
Comparison of next-generation sequencing platforms
|
Machine (manufacturer)
|
Chemistry
|
Modal read lengtha (bases)
|
Run time
|
Gb per run
|
Current, approximate cost (US$)b
|
Advantages
|
Disadvantages
|
|---|---|---|---|---|---|---|---|
|
High–end instruments
|
|||||||
|
454 GS FLX+ (Roche)
|
Pyrosequencing
|
700–800
|
23 h
|
0.7
|
500,000
|
Long read lengths
|
Appreciable hands-on time
|
|
High reagent costs
|
|||||||
|
High error rate in homopolymers
|
|||||||
|
HiSeq 2000/2500 (Illumina)
|
Reversible terminator
|
2 × 100
|
11 days (regular mode) or 2 days (rapid run mode)c
|
600 (regular mode) or 120 (rapid run mode)c
|
750,000
|
Cost-effectiveness
Steadily improving read lengths
Massive throughput
Minimal hands-on time
|
Long run time
Short read lengths
HiSeq 2500 instrument upgrade not available at time of writing (available end 2012)
|
|
5500xl SOLiD (Life Technologies)
|
Ligation
|
75 + 35
|
8 days
|
150
|
350,000
|
Low error rate
Massive throughput
|
Very short read lengths
Long run times
|
|
PacBio RS (Pacific Biosciences)
|
Real-time sequencing
|
3000 (maximum 15,000)
|
20 min
|
3 per day
|
750,000
|
Simple sample preparation
Low reagent costs
Very long read lengths
|
High error rate
Expensive system
Difficult installation
|
|
Bench–top instruments
|
|||||||
|
454 GS Junior (Roche)
|
Pyrosequencing
|
500
|
8 h
|
0.035
|
100,000
|
Long read lengths
|
Appreciable hands-on time
High reagent costs
High error rate in homopolymers
|
|
Ion Personal Genome Machine (Life Technologies)
|
Proton detection
|
100 or 200
|
3 h
|
0.01–0.1 (314 chip), 0.1–0.5 (316 chip), or up to 1 (318 chip)
|
80,000 (including OneTouch and server)
|
Short run times
Appropriate throughput for microbial applications
|
Appreciable hands-on time
High error rate in homopolymers
|
|
Ion Proton (Life Technologies)
|
Proton detection
|
Up to 200
|
2 h
|
Up to 10 (Proton I chip) or up to 100 (Proton II chip)
|
145,000 + 75,000 for compulsory server
|
Short run times
Flexible chip reagents
|
Instrument not available at time of writing
|
|
MiSeq (Illumina)
|
Reversible terminator
|
2 × 150
|
27 h
|
1.5
|
125,000
|
Cost-effectiveness
Short run times
Appropriate throughput for microbial applications
Minimal hands-on time
|
Read lengths too short for efficient assembly
|
10.2.4 Whole-Genome Shotgun Sequencing
Whole-genome shotgun sequencing (WGS) can provide highly accurate sequences in an economic way and has a fast turnaround (Hasan et al. 2014). WGS metagenomic sequencing has proved to be a powerful tool for studying the human microbiome. At present, WGS metagenomic data contain millions to billions of short reads and offer an unprecedented opportunity to identify species at or near strain level and their abundance.
10.2.5 Single-Cell Genome Sequencing
Remarkable in the identification of bacteria is single-cell genome sequencing which enables not only identification of microbes but links their functions to species, which is not feasible with metagenomic techniques. It also analyzes low-abundance species that can be lost in community-based analyses and can be useful in complementing metagenomic analyses (Yilmaz and Singh 2012). An ultimate goal of single-cell sequencing is recovery of genome sequences from each cell within an environment (Clingenpeel et al. 2015).
10.2.6 Metatranscriptomics of the Oral Microbiome during Health and Disease
Although new techniques have revealed what organisms are present in the oral microbiome, they do not tell anything about the viability of the organisms or their functions. Therefore efforts have been made recently to use microbiomics, metagenomics, and transcriptomics to better understand the role of the oral microbiome in health and disease. This may also help us to more efficiently prevent these diseases and provide a personalized treatment.
Our indigenous microbiota is closely linked to health. However, when disrupted the same microbiota can induce disease. Such diseases are characterized by changes in the relative amounts of different species. While such changes in the microbiota occur, it is also clear that the members of the microbial communities can differ markedly between individuals (Ge et al. 2013). This applies to the microbiota of both healthy and diseased individuals. In a study based on nine patient-matched healthy and diseased samples, 160,000 genes were compared in healthy and diseased periodontal communities (Jorth et al. 2014). Massive parallel RNA sequencing was used to demonstrate changes in the composition and gene expression of the microbiota in health and periodontitis. It was shown that both communities exhibited defined differences in metabolism that were conserved between patients. In contrast, the metabolic gene expression of individual species within the community varied greatly between patients. Disease-associated communities also showed conserved changes in metabolic and virulence gene expression. Thus, by using transcriptional profiling the authors could determine changes in the composition and gene expression of the human oral microbiota in health and periodontitis.
By using metatranscriptome analysis of periodontal biofilm in vitro, it was demonstrated that addition of periodontal pathogens to a healthy biofilm multispecies model had a drastic effect in changing the gene expression profiles of the organisms of the healthy community (Frias-Lopez and Duran-Pinedo 2012). Chaperones were highly upregulated, possibly due to stress, and there was a significant upregulation of ABC transporter systems and putative transposases. With pathogens present, proteins related to growth and division, as well as a large portion of transcription factors, were upregulated.
10.2.7 Community-Wide Transcriptome Analysis of the Oral Microbiome in Subjects With and Without Periodontitis
Our knowledge on the in situ activities of the organisms and their interaction with each other and with the environment is limited. Such knowledge may be obtained by characterizing gene expression profiles of the microbiome. In situ genome-wide transcriptome variation was studied in the subgingival microbiome of six periodontally healthy individuals and seven individuals with periodontitis (Duran-Pinedo et al. 2014). The overall metabolic activities defining disease were related to iron acquisition, lipopolysaccharide synthesis, and flagella synthesis. It was both noteworthy and unexpected that the majority of virulence factors upregulated in periodontitis came from organisms not considered as major pathogens. Also remarkable was that one of the organisms with characterized gene expression profile was from the uncultured candidate division TM7 exhibiting upregulation of putative virulence factors in disease. This demonstrated the importance of in situ metatranscriptomic studies for studying the possible etiological role of uncultured organisms. Unexpectedly, no viral sequence was detected in either the metagenome or the metatranscriptome.
10.3 Oral Microbiota in Health
The oral microbiota in health is highly diversified. It consists of approximately 600 predominant species (Dewhirst et al. 2010) that contribute to the health and physiology of the oral cavity. Two main types of tissues are colonized: soft and hard tissues. It is also clear that the oral cavity contains different niches for bacterial growth with different bacterial profiles that are site and subject specific (Fig. 10.2). Even close sites such as the dorsal and lateral sides of the tongue dorsum (Aas et al. 2005) and the vestibular and lingual surfaces of incisors and canines (Simon-Soro et al. 2013) have different microbiotas. The oral microbiota has, due to its continuum with the external environment, developed features to counteract challenges from foreign bacteria. There is probably a core microbiome for health which is common to all individuals (Zarco et al. 2012). In addition, there is a variable microbiome unique to individuals depending on lifestyle and physiological differences. Supporting the existence of a core microbiome was that identical bacterial sequences were detected in the oral cavities of unrelated healthy persons (Zaura et al. 2009). Transcription profiling defined a functional core microbiota of nearly 60 species in dental plaque (Peterson et al. 2014), and Wang et al. (2013) described a core disease-associated community in periodontitis by metagenomic sequencing. A study based on a large set of near full-length sequences in 10 healthy individuals identified 10 variables shared by 11 bacterial species (Bik et al. 2010). However, there were also significant interindividual differences. This supported the presence of both a core and a variable microbiome in the oral cavity. Based on several literature reports (Zarco et al. 2012) the major genera with the largest representation in the oral cavity were found to include Streptococcus, Veillonella, Granulicatella, Gemella, Actinomyces, Corynebacterium, Rothia, Fusobacterium, Porphyromonas, Prevotella, Capnocytophaga, Neisseria, Haemophilus, Treponema, Lactobacterium, Eikenella, Leptotrichia, Peptostreptococcus, Staphylococcus, Eubacteria, and Propionibacterium.
10.3.1 Microbiota in Periodontal Disease
Over the years, there have been several milestones and hypotheses on the microbial etiology of periodontitis (Hajishengallis and Lamont 2012). Etiologies related to specific organisms (amoeba, spirochetes, fusiforms, or streptococci), nonspecific plaque hypothesis/mixed anaerobic infections, microbial shift in periodontitis, specific plaque hypothesis, red complex bacteria (Porphyromonas gingivalis, Tannerella forsythia, Treponema denticola), ecological catastrophe hypothesis, disruption of periodontal tissue homeostasis, keystone pathogens, and polymicrobial synergy and dysbiosis (PSD) can be mentioned. This variability may partly be considered results of increased knowledge related to instrumental analytical improvements. However, rather than mentioning the microorganisms involved under each etiological heading, space will be devoted here to the most recent concept, PSD.
In the PSD model, it is recognized that the gingival crevice is colonized by a diverse microbiota where compatible microorganisms assemble into heterotypic communities. These are in equilibrium with the host. The organisms are controlled by the host, despite their production of toxic products such as proteases, overgrowth, and pathogenicity. Noteworthy, the microbial components of these communities vary over time from person to person and from site to site. The virulence of the entire community is increased by keystone pathogens such as P. gingivalis which can have interactive communication with accessory pathogens like the mitis group of streptococci, thereby orchestrating inflammatory disease by remodeling a normally benign microbiota into a dysbiotic one (Hajishengallis and Lamont 2012; Hajishengallis et al. 2012). The host immune response is not impaired and the abundance of the dysbiotic community increases, destroying tissue homeostasis and causing destruction of periodontal tissues. PSD is probably not the last model of periodontitis that will be launched, but it is attractive from the point that it reconciles the joint effects of a synergetic and a dysbiotic microbial community, rather than select organisms.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


