Periodontal Pathogenesis
Our knowledge of periodontal pathogenesis has evolved over the years. It is important to be aware of this, because treatment philosophies have similarly changed in parallel with our improving understanding of the disease processes. For example, during the late 1800s, Willoughby D. Miller (an eminent dental researcher who established the important causal role of oral bacteria for dental caries) asserted that “during the last few years the conviction has grown continually stronger, among physicians as well as dentists, that the human mouth, as a gathering-place and incubator of diverse pathogenic germs, performs a significant role in the production of varied disorders of the body, and that if many diseases whose origin is enveloped in mystery could be traced to their source, they would be found to have originated in the oral cavity.”116 This marked the beginning of an era of dental treatment strategies that aimed to treat systemic diseases by eliminating the so-called “foci of infection” in the mouth. As a result, many patients underwent unnecessary dental clearances to manage their systemic diseases.
By the 1930s, such approaches were beginning to be questioned as evidenced by a clinical study of 200 patients with rheumatoid arthritis of whom 92 had their tonsils removed as treatment for the arthritis (even though only about 15% gave any history of tonsillitis or sore throat) and of whom 52 had some or all of their teeth removed.28 Of the 92 who had their tonsils removed, there was no impact on the arthritis in 86 patients (although 2 got worse); of the 52 who had teeth removed, there was no benefit in 47 cases (and 3 reported a worsening of their arthritis after the extractions). The authors wrote that “focal infection is a splendid example of a plausible medical theory which is in danger of being converted by its too enthusiastic supporters into the status of an accepted fact.”28 The end of the focal infection era was signaled by an editorial in the Journal of the American Medical Association in 1952, which stated that “many patients with diseases presumably caused by foci of infection have not been relieved of their symptoms by removal of the foci, many patients with these same systemic diseases have no evident focus of infection, foci of infection are as common in apparently healthy persons as in those with disease.”165
Advances in the management of periodontitis have been driven by improved knowledge of the epidemiology, causation, and pathogenesis of the disease.195 During the 1970s, the role of plaque as the sole causative factor for periodontitis was unquestioned. In those days, nonsurgical treatment was in its infancy, and most treatment options involved surgery (e.g., gingivectomy for the treatment of shallower pockets, access flap surgery for the treatment of deeper sites). When looking back, it becomes clear that the treatment strategies used during a given time period are entirely dependent on the prevailing understanding of pathogenesis at that particular point in time. It is therefore very likely that the management options that we take for granted now will change again in the future. This is to be welcomed, because a progressive clinical discipline such as periodontology that is well founded in science and with patient benefit as its primary value should strive to improve therapeutic strategies in the light of continued discovery.
During the 1970s and 1980s, bacterial plaque was generally considered to be preeminent as the cause of periodontitis. At that time, it was accepted that poor oral hygiene resulted in increased plaque accumulation, which in turn resulted in periodontal disease. However, this model failed to take into account observations such as the fact that there are many individuals with poor oral hygiene who do not develop advanced periodontal disease and, conversely, that there are unfortunate individuals who, despite good oral hygiene and compliance with periodontal treatment protocols, continue to experience progressive periodontal breakdown and who would be considered to have aggressive periodontitis. These findings were confirmed by the work of Löe and colleagues, who studied Sri Lankan tea laborers who had no access to dental care and who could be divided into three main categories: (1) individuals (≈8% of the population studied) who had a rapid progression of periodontal disease; (2) those (≈81%) who had a moderate progression of such disease; and (3) those (≈11%) who demonstrated no progression of periodontal disease beyond gingivitis.107 All patients in this population displayed abundant plaque and calculus deposits. The causative role of plaque bacteria is clear in that the bacteria initiate and perpetuate the inflammatory responses that develop in the gingival tissues. However, the major determinant of susceptibility to disease is the nature of the immune–inflammatory responses themselves. It is paradoxical that these defensive processes, which are protective by intent (i.e., to prevent the ingress of the bacteria and their products into the tissues), result in the majority of tissue damage that leads to the clinical manifestations of disease.
Histopathology of Periodontal Disease
Clinically Healthy Gingival Tissues
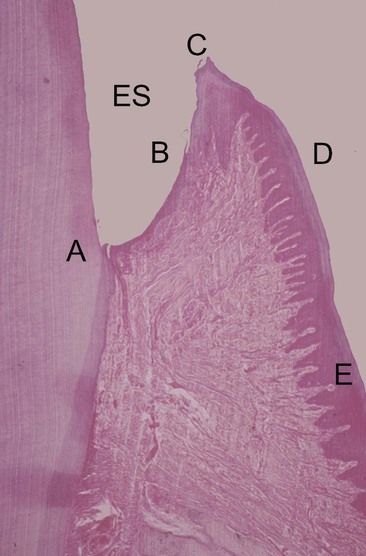
Clinically healthy gingival tissues (e.g., those observed in patients with excellent oral hygiene and no visible plaque deposits who typically have received regular and meticulous professional cleaning) are pink in appearance, not swollen, not inflamed, and firmly attached to the underlying tooth and bone, with minimal bleeding on probing. The dentogingival junction is a unique anatomic feature that functions to attach the gingiva to the tooth. It comprises an epithelial portion and a connective tissue portion, both of which are of fundamental importance for periodontal pathogenesis. The epithelial portion can be divided into three distinct epithelial structures: the gingival epithelium, the sulcular epithelium, and the junctional epithelium (Figure 5-1). These epithelial structures are in continuity with each other, but they have distinct structures and functions as indicated in Box 5-1.
The junctional epithelium is a particularly unique epithelial structure, because the surface cells are specialized for the purpose of attachment to the tooth.11 Therefore, unlike other epithelial tissues elsewhere in the body, there is no opportunity for the sloughing of cells from the surface. Instead, cells at the basal layer continually divide and move to within two or three cell layers of the tooth surface and then migrate coronally and parallel to the tooth surface to eventually reach the floor of the sulcus and then be sloughed off into the gingival crevice. The extracellular spaces between the junctional epithelium are also greater than those seen with other epithelial tissues, with the intercellular spaces comprising approximately 18% of the volume of the epithelium. This is a result of a lower density of desmosomes in the junctional epithelium as compared with the gingival epithelium; the junctional epithelium is therefore intrinsically “leaky.” This has great relevance for periodontal pathogenesis, because the widened intercellular spaces in the junctional epithelium permit the migration of neutrophils (polymorphonuclear leukocytes); they also allow macrophages from the gingival connective tissues to enter the sulcus to phagocytose bacteria, and the ingress of bacterial products and antigens occurs as well.
The connective tissue component of the dentogingival unit contains densely packed collagen fiber bundles (a mixture of type I and III collagen fibers) that are arranged in distinct patterns that maintain the functional integrity of the tissues and the tight adaptation of the soft tissues to the teeth. These include the following (see Chapter 1):
• Dentogingival fibers (extend from the cementum into the free and attached gingiva)
• Alveologingival fibers (extend from the alveolar crest into the free and attached gingiva)
• Circular fibers (wrap around the tooth, maintain the close adaptation of the free gingiva to the tooth, and interweave with other collagen fiber bundles)
• Dentoperiosteal fibers (run from the cementum over the alveolar crest and insert into the alveolar process)
• Transseptal fibers (run interdentally from the cementum just apical to the junctional epithelium and over the alveolar crest, where they insert into the cementum of the neighboring tooth).
It is important to note that, even in clinically healthy gingiva, the gingival connective tissue contains at least some inflammatory cells, particularly neutrophils. Neutrophils continually migrate through the connective tissues and pass through the junctional epithelium to enter the sulcus or pocket. These findings were reported in the classic investigations of the histology of periodontal disease reported by Page and Schroeder in 1976.132 This low-grade inflammation occurs in response to the continued presence of bacteria and their products in the gingival crevice. There is a continuous exudate of fluid from the gingival tissues that enters the crevice and flows out as gingival crevicular fluid (GCF). In addition to the continuous migration of neutrophils through the gingival tissues, lymphocytes and macrophages also accumulate. The presence of leukocytes in the connective tissues results from the chemotactic stimulus created by the subgingival biofilm and bacterial products as well as from the chemoattractant factors produced by the host.
• The maintenance of an intact epithelial barrier (the junctional and sulcular epithelium)
• The outflow of GCF from the sulcus (dilution effect and flushing action)
• The sloughing of surface epithelial cells of the junctional and sulcular epithelium
• The presence of neutrophils and macrophages in the sulcus that phagocytose bacteria
• The presence of antibodies in the GCF (although it is not clear whether these are effective)
However, if plaque accumulation increases so that these defense mechanisms are overwhelmed, then inflammation and the classic clinical signs of gingivitis will develop. Although the development of gingivitis in response to the accumulation of plaque is fairly predictable, research has identified that a spectrum of responses may be observed, with some individuals developing marked gingival inflammation for a given plaque challenge and others developing minimal gingival inflammation.183 These observations underscore the importance of variations in host responses between individuals in terms of gingival inflammatory responses. Furthermore, many individuals may never develop periodontitis, despite having widespread gingivitis. The host’s immune–inflammatory response is fundamental for determining which individuals may progress to developing periodontitis, and it is likely that inflammatory responses are markedly different in those individuals who develop periodontitis as compared with those who never progress beyond gingivitis. The challenge that this presents clinically is that we do not yet know enough about susceptibility to periodontitis to identify these individuals before they actually develop signs of disease.
Histopathology of Gingivitis and Periodontitis
The landmark studies of Page and Schroeder132 described the histologic changes that occur in the gingival tissues as the initial, early, established, and advanced gingival lesions. In broad terms, the initial lesion corresponds with clinically healthy (but nonetheless slightly inflamed) tissues, the early lesion corresponds with the early stages of (clinically evident) gingivitis, the established lesion corresponds with chronic gingivitis, and the advanced lesion marks the transition to periodontitis, with attachment loss and bone resorption. It is important to note that these are histologic descriptions only, and they should not form part of a clinical diagnosis. It is not possible to make any statements about the histologic status of a patient’s tissues, unless a biopsy is taken and the tissue examined microscopically. It is also important to note that these classic descriptions are primarily based on findings in experimental animals. The histologic stages of gingivitis are summarized in Box 5-2.
The Early Lesion.
The early lesion develops after about 1 week of continued plaque accumulation and corresponds with the early clinical signs of gingivitis. The gingiva are erythematous in appearance as a result of the proliferation of capillaries, the opening up of microvascular beds, and continued vasodilation.105 Increasing vascular permeability leads to increased GCF flow, and transmigrating neutrophils increase significantly in number. The predominant infiltrating cell types are neutrophils and lymphocytes (primarily thymic lymphocytes [T cells]),136 and the neutrophils migrate through the tissues to the sulcus and phagocytose bacteria. Fibroblasts degenerate, primarily via apoptosis (programmed cell death), which increases the space available for infiltrating leukocytes. Collagen destruction occurs, which results in collagen depletion in the areas apical and lateral to the junctional and sulcular epithelium. The basal cells of these epithelial structures begin to proliferate to maintain an intact barrier against the bacteria and their products, and the epithelium can then be seen proliferating into the collagen-depleted areas of the connective tissues (Figure 5-2).152 As a result of edema of the gingival tissues, the gingiva may appear slightly swollen, and, accordingly, the gingival sulcus becomes slightly deeper. The subgingival biofilm exploits this ecologic niche and proliferates apically (thereby rendering effective plaque control more difficult). The early gingival lesion may persist indefinitely, or it may progress further.
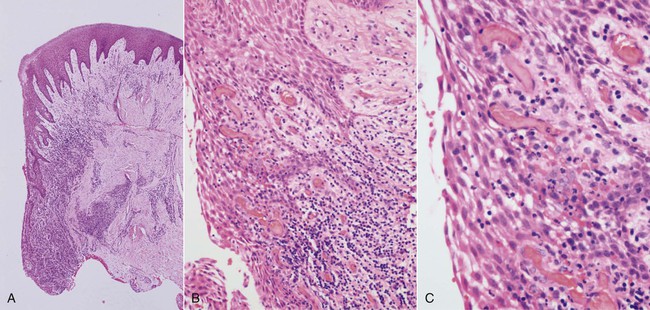
The Established Lesion.
The established lesion roughly corresponds with what clinicians would refer to as “chronic gingivitis.” The progression from the early lesion to the established lesion depends on many factors, including the plaque challenge (the composition and quantity of the biofilm), host susceptibility factors, and risk factors (both local and systemic). In the initial work by Page and Schroeder, the established lesion was defined as being dominated by plasma cells.132 In human studies, reports have suggested that plasma cells predominate in established gingivitis in older subjects,51 whereas lymphocytes predominate in younger individuals, although the relevance of these findings is not clear.23,51 What is clear from all of the studies is that there is a significant inflammatory cell infiltrate in established gingivitis that occupies a considerable volume of the inflamed connective tissues. Large numbers of infiltrating cells can be identified adjacent and lateral to the junctional and sulcular epithelium, around blood vessels, and between collagen fiber bundles.22 Collagen depletion continues, with further proliferation of the epithelium into the connective tissue spaces. Neutrophils accumulate in the tissues and release their lysosomal contents extracellularly (in an attempt to kill bacteria that are not phagocytosed), thereby resulting in further tissue destruction. Neutrophils are also a major source of matrix metalloproteinase-8 (MMP-8; neutrophil collagenase) and MMP-9 (gelatinase B), and these enzymes are produced in large quantities in the inflamed gingival tissues as the neutrophils migrate through the densely packed collagen fiber bundles to enter the sulcus. The junctional and sulcular epithelium form a pocket epithelium that is not firmly attached to the tooth surface, that contains large numbers of neutrophils, and that is more permeable to the passage of substances into or out of the underlying connective tissue. The pocket epithelium may be ulcerated and less able to resist the passage of the periodontal probe, so bleeding on probing is a common feature of chronic gingivitis. It is important to remember that these inflammatory changes are still completely reversible if effective plaque control is reinstituted.
The Advanced Lesion.
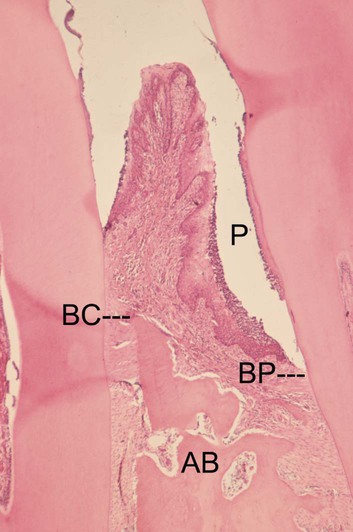
The advanced lesion marks the transition from gingivitis to periodontitis. This transition is determined by many factors, the relative importance of which is currently unknown but which includes the bacterial challenge (both the composition and the quantity of the biofilm), the host inflammatory response, and susceptibility factors, including environmental and genetic risk factors. Histologic examination reveals continued evidence of collagen destruction that extends into the periodontal ligament and the alveolar bone. Neutrophils predominate in the pocket epithelium and the periodontal pocket, and plasma cells dominate in the connective tissues. The junctional epithelium migrates apically along the root surface into the collagen-depleted areas to maintain an intact epithelial barrier. Osteoclastic bone resorption commences, and the bone retreats from the advancing inflammatory front as a defense mechanism to the prevent spread of bacteria into the bone (Figure 5-3). As the pocket deepens, plaque bacteria proliferate apically into a niche, which is very favorable for many of the species that are regarded as periodontal pathogens. The pocket presents a protected, warm, moist, and anaerobic environment with a ready nutrient supply, and, because the bacteria are effectively outside of the body (even though they are in the periodontal pocket), they are not significantly eliminated by the inflammatory response. Thus, a cycle develops in which chronic inflammation and associated tissue damage continue. The tissue damage is mainly caused by the inflammatory response, yet the initiating factor—the biofilm—is not eliminated. The destruction of collagen fibers in the periodontal ligament continues, bone resorption progresses, the junctional epithelium migrates apically to maintain an intact barrier, and as a result, the pocket deepens fractionally. This makes it even more difficult to remove the bacteria and to disrupt the biofilm through oral hygiene techniques, and thus the cycle is perpetuated.
Inflammatory Responses in the Periodontium
Microbial Virulence Factors
Lipopolysaccharide.
Lipopolysaccharides (LPSs) are large molecules composed of a lipid component (lipid A) and a polysaccharide component. They are found in the outer membrane of gram-negative bacteria, they act as endotoxins (LPSs are frequently referred to as endotoxins), and they elicit strong immune responses in animals. LPSs are highly conserved in gram-negative bacterial species, which reflects their importance in maintaining the structural integrity of the bacterial cells. Immune systems in animals have evolved to recognize LPS via Toll-like receptors (TLRs), a family of cell surface molecules that are highly conserved in animal species ranging from Drosophila (a genus of fruit flies) to humans, thereby reflecting their importance in innate immune responses. TLRs are also present in lower animals and are in fact more varied than in higher species.26 TLRs are cell surface receptors that recognize microbe-associated molecular patterns (MAMPs), which are conserved molecular structures located on diverse pathogens. TLR-4 recognizes LPSs from gram-negative bacteria and functions as part of a complex of cell surface molecules, including CD14 and MD-2 (also known as lymphocyte antigen 96). The interaction of this CD14/TLR-4/MD-2 complex with LPSs triggers a series of intracellular events, the net result of which is the increased production of inflammatory mediators (most notably cytokines) and the differentiation of immune cells (e.g., dendritic cells) for the development of effective immune responses against the pathogens. It is particularly interesting to the periodontist that the pathogen Porphyromonas gingivalis has an atypical form of LPSs that are recognized by both TLR-2 and TLR-4.38,45
Bacterial Enzymes and Noxious Products.
Plaque bacteria produce a number of metabolic waste products that contribute directly to tissue damage. These include noxious agents such as ammonia (NH3) and hydrogen sulfide (H2S) as well as short-chain carboxylic acids such as butyric acid and propionic acid. These acids are detectable in GCF and found in increasing concentrations as the severity of periodontal disease increases. These substances have profound effects on host cells (e.g., butyric acid induces apoptosis in T cells, B cells, fibroblasts, and gingival epithelial cells).95,96,166 The short-chain fatty acids may aid P. gingivalis infection through tissue destruction, and they may also create a nutrient supply for the organism by increasing bleeding into the periodontal pocket. The short-chain fatty acids also influence cytokine secretion by immune cells, and they may potentiate inflammatory responses after exposure to proinflammatory stimuli such as LPS, interleukin-1β (IL-1β), and tumor necrosis factor alpha (TNF-α).123
Plaque bacteria produce proteases, which are capable of breaking down structural proteins of the periodontium such as collagen, elastin, and fibronectin. Bacteria produce these proteases to digest proteins and thereby provide peptides for bacterial nutrition. Bacterial proteases disrupt host responses, compromise tissue integrity, and facilitate the microbial invasion of the tissues. P. gingivalis produces two classes of cysteine proteases that have been implicated in periodontal pathogenesis. These are known as gingipains, and they include the lysine-specific gingipain Kgp and the arginine-specific gingipains RgpA and RgpB. The gingipains can modulate the immune system and disrupt immune–inflammatory responses, potentially leading to increased tissue breakdown.138 Gingipains can reduce the concentrations of cytokines in cell culture systems,7 and they digest and inactivate TNF-α.25 The gingipains can also stimulate cytokine secretion via the activation of protease-activated receptors (PARs). For example, RgpB activates two different PARs (PAR-1 and PAR-2), thereby stimulating cytokine secretion,108 and both Rgp and Kgp gingipains stimulate IL-6 and IL-8 secretion by monocytes via the activation of PAR-1, PAR-2 and PAR-3.184
Microbial Invasion.
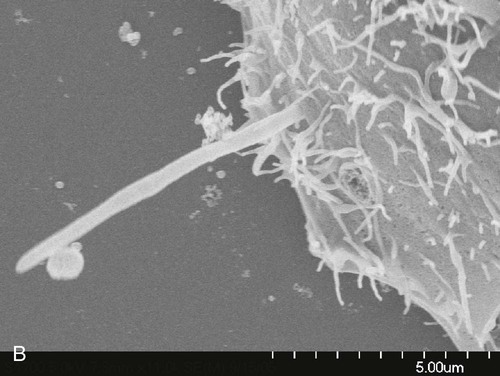
Microbial invasion of the periodontal tissues has long been a contentious topic. In histologic specimens, bacteria (including cocci, filaments, and rods) have been identified in the intercellular spaces of the epithelium.50 Periodontal pathogens such as P. gingivalis and Aggregatibacter actinomycetemcomitans have been reported to invade the gingival tissues,30,76,148 including the connective tissues.147 Fusobacterium nucleatum can invade oral epithelial cells, and bacteria that routinely invade host cells may facilitate the entry of noninvasive bacteria by coaggregating with them (Figure 5-4).47 It has also been shown that A. actinomycetemcomitans can invade epithelial cells and persist intracellularly.49 The clinical relevance of these findings is unclear, however. Some investigators have suggested that tissue invasion by subgingival bacteria is an active process, whereas others have considered it to be an artifact or simply a passive translocation process.

Fimbriae.
The fimbriae of certain bacterial species, particularly P. gingivalis, may also play a role in periodontal pathogenesis. P. gingivalis fimbriae stimulate immune responses, such as IL-6 secretion,97,128 and the major fimbrial structural component of P. gingivalis, FimA, has been shown to stimulate nuclear factor (NF)-κβ and IL-8 in a gingival epithelial cell line via TLR-2.5. Monocytes are also stimulated by P. gingivalis FimA, secreting IL-6, IL-8, and TNF-α.48 P. gingivalis fimbriae also interact with complement receptor-3 (CR-3) to activate intracellular signaling pathways that inhibit IL-12 production mediated by TLR-2 signalling.66 This may be of clinical relevance, because IL-12 is important in the activation of natural killer (NK) cells and CD8+ cytotoxic T cells, which themselves may be important in killing P. gingivalis–infected host cells, such as epithelial cells. Indeed, the blockade of the CR-3 receptor promotes IL-12–mediated clearance of P. gingivalis and negates its virulence.66 Bacterial fimbriae are therefore important for modifying and stimulating immune responses in the periodontium.
Bacterial Deoxyribonucleic Acid and Extracellular Deoxyribonucleic Acid.
Bacterial deoxyribonucleic acid (DNA) stimulates immune cells via TLR-9, which recognizes hypomethylated CpG regions of the DNA.93 CpG sites are regions of DNA at which a cytosine nucleotide is found next to a guanine nucleotide (separated by a phosphate molecule, which links the C and G nucleotides together, hence “CpG”). Extracellular DNA (eDNA) is likely to play a role in the development and structure of the biofilms formed by oral bacteria, and it has been identified as an important component of the matrix in a number of bacterial biofilms.169,196 eDNA is derived from the chromosomal DNA of bacteria in biofilms, and the majority of eDNA is released after bacterial cell lysis.2,178 However, there is also evidence that eDNA secretion may occur from bacterial cells by mechanisms that are independent of cell lysis.68,142 The significance of this finding is not yet clear, but such “donated” DNA may be used by bacterial species as a means of increasing genetic diversity (if taken up by other bacteria), thereby contributing to antigenic variation and the spread of antibiotic resistance, and it may also modulate the host immune response. Thus eDNA may function as a source of genetic information for naturally transformable bacteria in the biofilm191 or as a stimulus for host immunity. Little is known about the role of eDNA in oral biofilms, however. It has been demonstrated that DNA isolated from P. gingivalis, A. actinomycetemcomitans, and Peptostreptococcus micros stimulates macrophages and gingival fibroblasts to produce TNF-α and IL-6 in a dose-dependent manner; therefore, immune stimulation by bacterial DNA from subgingival species could contribute to periodontal pathogenesis.124
Host-Derived Inflammatory Mediators
Cytokines.
Cytokines play a fundamental role in inflammation, and they are key inflammatory mediators in periodontal disease.140,161 They are soluble proteins, and they act as messengers to transmit signals from one cell to another. Cytokines bind to specific receptors on target cells, and they initiate intracellular signaling cascades that result in phenotypic changes in the cell via altered gene regulation.17,176 Cytokines are effective in very low concentrations, they are produced transiently in the tissues, and they primarily act locally in the tissues in which they are produced. Cytokines are able to induce their own expression in either an autocrine or paracrine fashion, and they have pleiotropic effects (i.e., multiple biologic activities) on a large number of cell types. (Autocrine signaling means that the autocrine agent [in this case cytokines] binds to receptors on the cell that secreted the agent, whereas paracrine signaling affects other nearby cells.) Simply put, cytokines bind to cell surface receptors and trigger a sequence of intracellular events that lead ultimately to the production of protein by the target cell, which alters that cell’s behavior and could result in, for example, the increased secretion of more cytokines in a positive feedback cycle that leads to inflammation.
Cytokines are produced by a large number of cell types, including infiltrating inflammatory cells (e.g., neutrophils, macrophages, lymphocytes) as well as resident cells in the periodontium (e.g., fibroblasts, epithelial cells).172 Cytokines signal, broadcast, and amplify immune responses, and they are fundamentally important for regulating immune–inflammatory responses and for combating infections. However, they also have profound biologic effects that lead to tissue damage with chronic inflammation; the prolonged and excessive production of cytokines and other inflammatory mediators in the periodontium leads to the tissue damage that characterizes the clinical signs of the disease. For example, cytokines mediate connective tissue and alveolar bone destruction through the induction of fibroblasts and osteoclasts to produce proteolytic enzymes (i.e., MMPs) that break down structural components of these connective tissues.12
There is significant overlap and redundancy between the function of individual cytokines. Cytokines do not act in isolation; rather, they function in flexible and complex networks that involve both proinflammatory and anti-inflammatory effects and that bring together aspects of both innate and acquired immunity.8 Cytokines play a key role at all stages of the immune response in periodontal disease.140 Among the most studied (and probably the most important) cytokines in periodontal pathogenesis are the proinflammatory cytokines IL-1β and TNF-α. Both of these cytokines play a key role in the initiation, regulation, and perpetuation of innate immune responses in the periodontium, thereby resulting in vascular changes and the migration of effector cells such as neutrophils into the periodontium as part of a normal immune response to the presence of subgingival bacteria.57
Matrix Metalloproteinases.
MMPs are a family of proteolytic enzymes that degrade extracellular matrix molecules such as collagen, gelatin, and elastin. They are produced by a variety of cell types, including neutrophils, macrophages, fibroblasts, epithelial cells, osteoblasts, and osteoclasts. The names and functions of key MMPs are shown in Table 5-1. The nomenclature of MMPs has been based on the perception that each enzyme has its own specific substrate; for example, MMP-8 and MMP-1 are both collagenases (i.e., they break down collagen). However, it is now appreciated that MMPs usually degrade multiple substrates, with significant substrate overlap between individual MMPs.70 The substrate-based classification is still used, however, and MMPs can be divided into collagenases, gelatinases/type IV collagenases, stromelysins, matrilysins, membrane-type metalloproteinases, and others.
TABLE 5-1
Classification of Matrix Metalloproteinases
| Group | Enzyme | Name |
| Collagenases | MMP-1 | Collagenase 1, fibroblast collagenase |
| MMP-8 | Collagenase 2, neutrophil collagenase | |
| MMP-13 | Collagenase 3 | |
| Gelatinases | MMP-2 | Gelatinase A |
| MMP-9 | Gelatinase B | |
| Stromelysins | MMP-3 | Stromelysin 1 |
| MMP-10 | Stromelysin 2 | |
| MMP-11 | Stromelysin 3 | |
| Matrilysins | MMP-7 | Matrilysin 1, pump-1 |
| MMP-26 | Matrilysin 2 | |
| Membrane-type MMPs | MMP-14 | MT1-MMP |
| MMP-15 | MT2-MMP | |
| MMP-16 | MT3-MMP | |
| MMP-17 | MT4-MMP | |
| MMP-24 | MT5-MMP | |
| MMP-25 | MT6-MMP | |
| Others | MMP-12 | Macrophage elastase |
| MMP-19 | — | |
| MMP-20 | Enamelysin |
MMPs, Matrix metalloproteinases; MT, membrane type.
(Adapted from Hannas AR, Pereira JC, Granjeiro JM, et al: Acta Odontol Scand 65:1-13, 2007).
MMPs are secreted in a latent form (inactive) and are activated by the proteolytic cleavage of a portion of the latent enzyme. This is achieved by proteases, such as cathepsin G, produced by neutrophils. MMPs are inhibited by proteinase inhibitors, which have anti-inflammatory properties. Key inhibitors of MMPs found in the serum include the glycoprotein α1-antitrypsin and α2-macroglobulin, a large plasma protein produced by the liver that is capable of inactivating a wide variety of proteinases. Inhibitors of MMPs that are found in the tissues include the tissue inhibitors of metalloproteinases (TIMPs), which are produced by many cell types; the most important in periodontal disease is TIMP-1.18 MMPs are also inhibited by the tetracycline class of antibiotics, which has led to the development of a subantimicrobial formulation of doxycycline as a licensed systemic adjunctive drug treatment for periodontitis that exploits the anti-MMP properties of this molecule (see Chapter 50).
Role of Specific Inflammatory Mediators in Periodontal Disease
Interleukin-1 Family Cytokines.
The IL-1 family of cytokines comprises at least 11 members, including IL-1α, IL-1β, IL-1 receptor antagonist (IL-1Ra), IL-18, and IL-33.140
IL-1β plays a key role in inflammation and immunity; it is closely linked to the innate immune response, and it induces the synthesis and secretion of other mediators that contribute to inflammatory changes and tissue damage. For example, IL-1β stimulates the synthesis of PGE2, platelet-activating factor, and nitrous oxide, thereby resulting in vascular changes associated with inflammation and increasing blood flow to the site of infection or tissue injury. IL-1β is mainly produced by monocytes, macrophages, and neutrophils and also by other cell types such as fibroblasts, keratinocytes, epithelial cells, B cells, and osteocytes.40 IL-1β increases the expression of ICAM-1 on endothelial cells and stimulates the secretion of the chemokine CXCL8 (which is IL-8), thereby stimulating and facilitating the infiltration of neutrophils into the affected tissues. IL-1β also synergizes with other proinflammatory cytokines and PGE2 to induce bone resorption. IL-1β has a role in adaptive immunity; it regulates the development of antigen-presenting cells (APCs) (e.g., dendritic cells), stimulates IL-6 secretion by macrophages (which in turn activates B cells), and has been shown to enhance the antigen-mediated stimulation of T cells.13 GCF concentrations of IL-1β are increased at sites affected by gingivitis73 and periodontitis,99 and tissue levels of IL-1β correlate with clinical periodontal disease severity.167 Studies in experimental animals have shown that IL-1β exacerbates inflammation and alveolar bone resorption.88 It is clear from the multiplicity of studies that have investigated this cytokine that IL-1β plays a fundamental role in the pathogenesis of periodontal disease.91
IL-1α is primarily an intracellular protein that is not normally secreted and that therefore is not usually found in the extracellular environment or in the circulation.43 Unlike IL-1β, biologically active IL-1α is constitutively expressed and likely mediates inflammation only when it is released from necrotic cells, thus acting as an “alarmin” to signal the immune system during cell and tissue damage.16 The precise role of IL-1α in periodontal pathogenesis is not well defined, although studies have reported elevated IL-1α levels in GCF and gingival tissues in patients with periodontitis.141 IL-1α is a potent bone-resorbing factor involved in the bone loss that is associated with inflammation.174 It is possible that the measured level of IL-1α in gingival tissues represents intracellular IL-1α that has been released from damaged or necrotic cells. It is probable that IL-1α plays a role in periodontal pathogenesis, possibly as a signaling cytokine (signaling tissue damage) and contributing to bone resorptive activity.
IL-1Ra has structural homology to IL-1β, and it binds to the IL-1 receptor (IL-1R1). However, the binding of IL-1Ra does not result in signal transduction; therefore, IL-1Ra antagonizes the action of IL-1β.42 IL-1Ra is important for the regulation of inflammatory responses, and it can be considered to be an anti-inflammatory cytokine. IL-1Ra levels have been reported to be elevated in the GCF and tissues of patients with periodontal disease, thereby suggesting that it has a role in immunoregulation in cases of periodontitis.144
IL-18 interacts with IL-1β and shares many of the pro-inflammatory effects of IL-1β.140 It is mainly produced by stimulated monocytes and macrophages.63 There is increasing evidence to suggest that IL-18 plays a significant role in inflammation and immunity. IL-18 results in pro-inflammatory responses, including the activation of neutrophils.102 It is a chemoattractant for T cells,89 and it interacts with IL-12 and IL-15 to induce interferon gamma (IFN-γ), thereby inducing T-helper (Th1) cells, which activate cell-mediated immunity.199 Interestingly, in the absence of IL-12, IL-18 induces IL-4 and a Th2 response, which regulates humoral (antibody-mediated) immunity.200 There is very limited direct evidence for a role of IL-18 in periodontal pathogenesis. Oral epithelial cells secrete IL-18 in response to stimulation with LPS,145 and a correlation between GCF IL-18 levels and sulcus depth has been reported.82 IL-18 levels have been reported to be higher than those of IL-1β in patients with periodontitis, thereby suggesting that IL-18—along with IL-1β—is predominant in periodontitis lesions.130 Because IL-18 has the ability to induce either Th1 or Th2 differentiation, it is likely to play an important role in periodontal disease pathogenesis.131
Other Interleukin-1 Family Cytokines.
Six new members of the IL-1 family (IL-1F) of cytokines have been identified on the basis of their sequence homology, structure, gene location, and receptor binding.4,10 Several of these cytokines were identified by different groups, who gave them a variety of names, and proposals were suggested for renaming all of the IL-1F cytokines in a more consistent manner, as indicated in Table 5-2. Our knowledge of the role of these cytokines in inflammation and immunity is very limited at present, and some of these cytokines may be evolutionarily redundant. IL-1F6, IL-1F8, and IL-1F9 are potential agonists (stimulating proinflammatory responses),19,182 whereas IL-1F5 and IL-1F10 are potential antagonists.19,33,103 IL-1F7 appears to have anti-inflammatory action.44 It has five splice variants and one isoform, IL-1F7b, which is highly expressed by monocytes and upregulated by LPS.24 An intracellular mode of action has been suggested for IL-1F7b; it translocates to the nucleus of macrophages, and it may act as a transcriptional modulator by reducing the production of LPS-stimulated proinflammatory cytokines, thus supporting an anti-inflammatory role for this cytokine.163
TABLE 5-2
Nomenclature of Interleukin-1 Family Cytokines
| Cytokine | Systematic Name | Function |
| IL-1α | IL-1F1 | Intracellular protein, pro-inflammatory, contributes to bone resorption, functions as an intracellular transcriptional regulator |
| IL-1β | IL-1F2 | Key role in inflammation and innate immunity, synergizes with other pro-inflammatory mediators, major role in adaptive immunity (i.e., regulation of T cells and myeloid cells), stimulates connective tissue breakdown and bone resorption |
| IL-1Ra | IL-1F3 | Inhibits the action of IL-1α and IL-1β |
| IL-18 | IL-1F4 | Similar pro-inflammatory profile to IL-1β, activates neutrophils, synergizes with IL-12 to activate T-helper 1 cells |
| IL-1F5 | IL-1F5 | Anti-inflammatory effects via IL-4 induction, antagonizes IL-1F6 action |
| IL-1F6 | IL-1F6 | Pro-inflammatory but restricted expression (e.g., localized to skin) |
| IL-1F7 | IL-1F7 | Anti-inflammatory, acts as an intracellular regulator, reduces production of lipopolysaccharide-stimulated pro-inflammatory cytokines |
| IL-1F8 | IL-1F8 | Pro-inflammatory but restricted expression (e.g., localized to skin and synovial tissues) |
| IL-1F9 | IL-1F9 | Pro-inflammatory but restricted expression (e.g., localized to skin, placenta, and esophagus) |
| IL-1F10 | IL-1F10 | Putative antagonist with anti-inflammatory action. |
| IL-33 | IL-1F11 | Activation of T-helper 2 cells and mast cells, functions as an intracellular transcriptional regulator but restricted expression (e.g., endothelial cells, smooth muscle cells, and fibroblasts) |
These novel IL-1F cytokines have limited tissue expression. For example, the agonists IL-1F6, IL-1F8, and IL-1F9 are mainly expressed in skin.182 Therefore, although the primary cellular sources of IL-1β and IL-18 are hematopoietic cells (e.g., neutrophils, macrophages, monocytes, lymphocytes), IL-1F5 through IL-1F10 are mainly expressed outside of these lineages. At present, there are no data to support a role for IL-1F5 through IL-1F10 in periodontal pathogenesis. However, given that they are expressed mainly by epithelial cells, it will be interesting to learn whether they may play a role in inflammatory responses in the gingiva. This is relevant given the continual exposure of gingival epithelial cells to bacterial challenge, and these cytokines also have properties similar to the primary cytokines (e.g., IL-1β). For example, LPS results in the upregulation of IL-1F6, IL-1F8, and IL-1F9, and these cytokines also stimulate the secretion of IL-6 and IL-8.182 P. gingivalis LPS upregulates IL-1F9 mRNA expression in monocytes, although it does not have an effect on IL-1F6, IL-1F7, IL-1F8, or IL-1F10.10
IL-33, which is also known as IL-1F11, is of particular interest because, uniquely among the IL-1 cytokines, it stimulates the production of Th2 cytokines (e.g., IL-5, IL-13), it activates Th2 cells, and it plays a role in mast cell development and function.1,77,90,117,151 IL-33 is mainly found in nonimmune cells such as bronchial and arterial smooth muscle cells and epithelial cells from the bronchus.151 It is constitutively expressed in the endothelial cells of small and large blood vessels, in the fibroblastic reticular cells of lymphoid tissues, and in epithelial cells.27,118 Our knowledge of the expression of IL-33 in myeloid immune cells is very limited, and there are no data to support a role for IL-33 in periodontal pathogenesis. However, it has been reported that IL-33 activates Th2 cells151 and that it is chemoattractant for these cells.90 Given that Th2 cells are likely to play a role in the destructive phases of periodontal disease and that the balance of T-cell subsets is an important factor in determining disease progression,58 IL-33 may yet prove to play a role in periodontal pathogenesis.
Tumor Necrosis Factor-α.
TNF-α is a key inflammatory mediator in periodontal disease, and it shares many of the cellular actions of IL-1β.64 It plays a fundamental role in immune responses, it increases neutrophil activity, and it mediates cell and tissue turnover by inducing MMP secretion. TNF-α stimulates the development of osteoclasts and limits tissue repair via the induction of apoptosis in fibroblasts. TNF-α is secreted by activated macrophages as well as by other cell types, particularly in response to bacterial LPS. The proinflammatory effects of TNF-α include the stimulation of endothelial cells to express selectins that facilitate leukocyte recruitment, the activation of macrophage IL-1β production, and the induction of PGE2 by macrophages and gingival fibroblasts.134 TNF-α—although it possesses similar activity to IL-1β—has a less potent effect on osteoclasts, and it is present at lower levels in inflamed gingival tissues than IL-1β.168 GCF levels of TNF-α increase as gingival inflammation develops, and higher levels are found in individuals with periodontitis.64,73 The importance of TNF-α (and IL-1β) in periodontal pathogenesis is unquestioned, and it has particularly been highlighted by studies showing that the application of antagonists to IL-1β and TNF-α resulted in an 80% reduction in recruitment of inflammatory cells in proximity to the alveolar bone and a 60% reduction in bone loss.6
Interleukin-6 and Related Cytokines.
The cytokines in this group—which include IL-6, IL-11, leukemia-inhibitory factor (LIF), and oncostatin M—share common signaling pathways via signal transducers glycoprotein (gp) 130.74 IL-6 is the most extensively studied of this group, and it has pleiotropic proinflammatory properties.87 IL-6 secretion is stimulated by cytokines such as IL-1β and TNF-α, and it is produced by a range of immune cells (e.g., T cells, B cells, macrophages, dendritic cells) as well as resident cells (e.g., keratinocytes, endothelial cells, fibroblasts).188 IL-6 is also secreted by osteoblasts, and it stimulates bone resorption and the development of osteoclasts.81,94 IL-6 is elevated in the cells, tissues, and GCF of patients with periodontal disease.56,104 IL-6 may have an influence on monocyte differentiation into osteoclasts and a role in bone resorption in patients with periodontal disease.129 IL-6 also has a key role in regulating the proliferation and differentiation of B cells and T cells, particularly the Th17 subset.87 IL-6 therefore has an important role in periodontal pathogenesis, although it is less than that of IL-1β or TNF-α.
IL-6 also has many activities outside of the immune system, such as in the cardiovascular and nervous systems. It has an important role in hematopoiesis and in signaling the production of C-reactive protein in the liver. Furthermore, IL-6 stimulates T-cell differentiation and function, and it is important in the regulation of the balance of T-cell subsets, particularly the activation of Th17 cells (a subset of T cells that produce IL-17) and the balance with regulatory T cells (Treg cells).
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


