CHAPTER 42 Antineoplastic Drugs
HISTORY OF CANCER CHEMOTHERAPY
The cytotoxic effects of drugs were observed well before the turn of the twentieth century, but their usefulness in the treatment of disease was not appreciated until the mid-1940s. Chemical warfare with sulfur mustard gas in World War I resulted in shrinkage of lymph nodes and myeloid tissues in the victims. The application of these nitrogen mustard compounds for the medical treatment of Hodgkin’s disease, malignant lymphomas, and chronic leukemia followed these observations but was not reported until the end of World War II. In 1944, glucocorticoids were shown to have a profound effect on the volume, structure, and function of lymphoid tissue.17 Subsequently, this effect was used in the control of human leukemia, and since then prednisone and prednisolone have been incorporated in drug protocols designed to ablate lymphoproliferative and myeloproliferative diseases.
In 1948, Farber and colleagues20 obtained temporary remissions in children with acute leukemia who were given the folic acid antagonist 4-aminopteroylglutamic acid (aminopterin). This specially tailored molecule was the first antimetabolite to produce unequivocally beneficial results in a human neoplastic disease.
The first antibiotic with activity against human tumors was actinomycin D. Introduced as an anticancer agent in 1952, dactinomycin (actinomycin D) is curative in many patients with Wilms’ tumor and uterine choriocarcinoma. The anticancer effects of the vinca alkaloids, extracted from the periwinkle plant (Vinca rosea), were initially shown in animals with experimental leukemia in 1960.40 In the same year, vinblastine was found to be valuable in the treatment of acute forms of leukemia, Hodgkin’s disease, and adenocarcinoma of the colon.35 The earliest reports of the use of carmustine, the prototype of the nitrosourea group of cytotoxic compounds, against human malignancies appeared in 1966.
In 1967, the enzyme l-asparaginase was found to produce remissions in some patients with acute leukemia. The first of the heavy metal complexes to have significant success in the treatment of human cancer was cisplatin, introduced in 1969. The 1950s and 1960s brought rapid development of new agents, and continued refinements in their use occurred in the 1970s and early 1980s with additional combination chemotherapy regimens and a better understanding of the cytokinetics of tumor cells and the pharmacokinetics of the drugs.13,15 The late 1980s and early 1990s contributed several new agents, such as taxanes, topoisomerase I inhibitors, and others with measurable efficacy and decreased toxicity; biologic response modifiers such as interferon and interleukin-2; and chemoprotective agents and newer technologies for the application of these antineoplastic agents.
The late 1990s brought the commercial availability of some MAbs for the treatment of several cancers, as well as important research on the role of angiogenesis, which had started in the 1960s. Angiogenesis, which is the formation of new blood vessels, plays a role in supporting existing tumors with required nutrients and oxygen and in forming metastatic tumors. The identification of angiogenic factors such as VEGF, basic fibroblastic growth factor, and other regulators and inhibitors of angiogenesis is leading to the development of new drugs to target these factors and evaluate their role in starving cancer cells and preventing the formation of metastatic disease.44
PRINCIPLES OF CANCER CHEMOTHERAPY
Chemotherapy drugs kill or impair susceptible tumor cells by blocking a drug-sensitive biochemical or metabolic pathway. Some, such as cell cycle phase–specific antimetabolites, act by inhibiting DNA synthesis and are most effective against rapidly dividing cells. Others, including alkylating agents, act by interfering with nucleic acid function and protein production throughout the cell division cycle and are effective against both proliferating and resting cells (Figures 42-1 and 42-2). All chemotherapy drugs are extremely cytotoxic with low margins of safety. Incorporating the current understanding of tumor biology, the patient’s physiologic status, and the drug’s pharmacologic features, the principles that govern the useful application of cancer chemotherapy include the following:
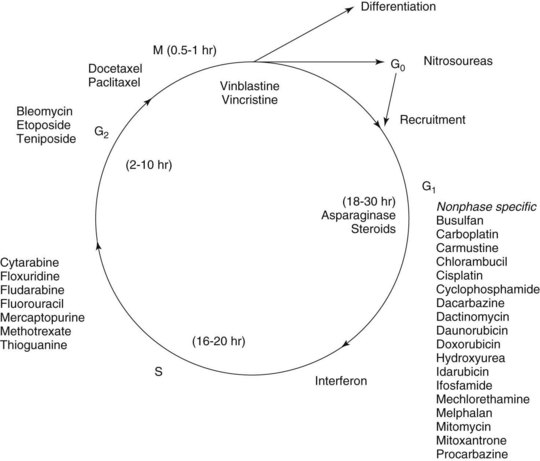
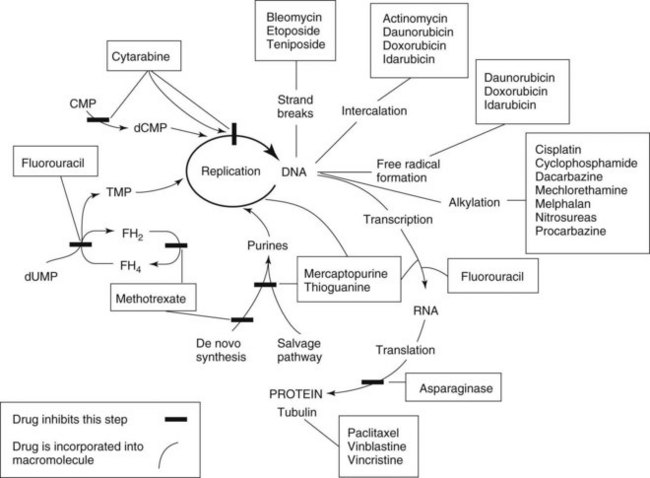
CHEMOTHERAPEUTIC DRUGS
Antineoplastic Alkylating Agents
Alkylating agents (Table 42-1) are composed of six major chemical classes: (1) nitrogen mustards (chlorambucil, cyclophosphamide, estramustine, ifosfamide, mechlorethamine, and melphalan), (2) alkyl sulfonates (busulfan), (3) ethylenimines (thiotepa), (4) triazines (dacarbazine), (5) tetrazines (temozolomide), and (6) nitrosoureas (carmustine, lomustine, and streptozocin). They all share the common chemical characteristic of forming alkyl radicals, which form covalent linkages with nucleophilic moieties such as the phosphate, sulfhydryl, hydroxyl, carboxyl, amino, and imidazole groups. This radical formation allows them to react with organic compounds such as DNA and RNA and proteins essential for cell metabolism and protein synthesis. By binding these groups, they also prevent cell division by cross-linking strands of DNA.
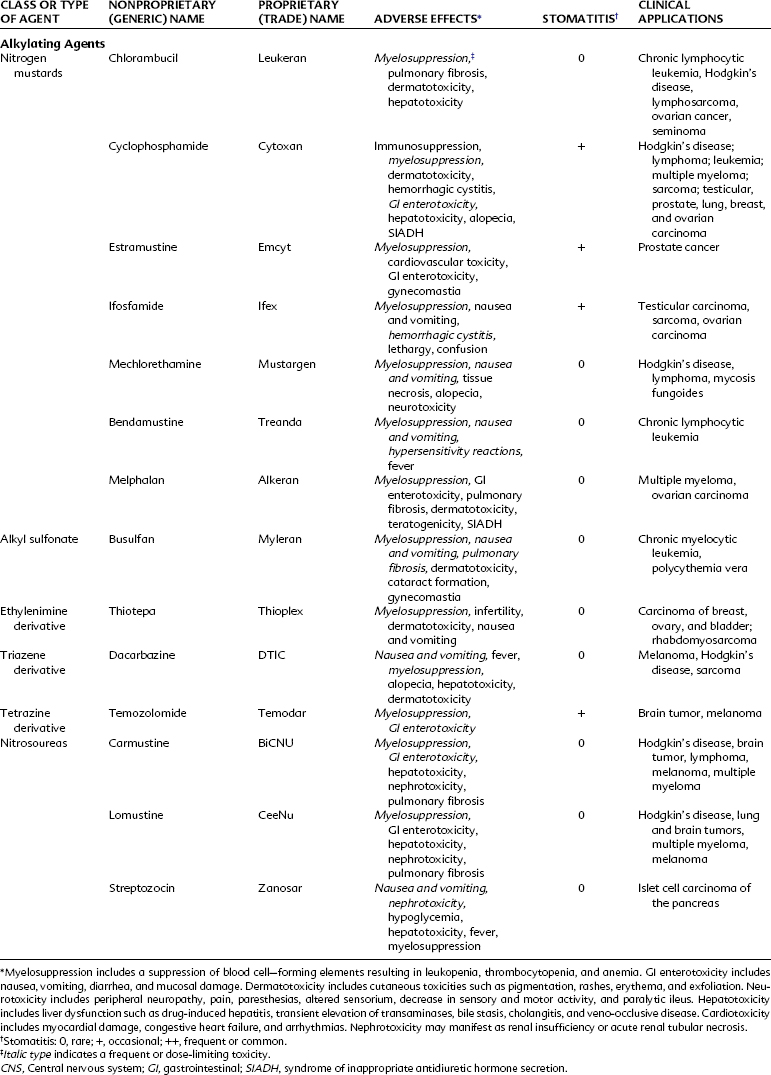
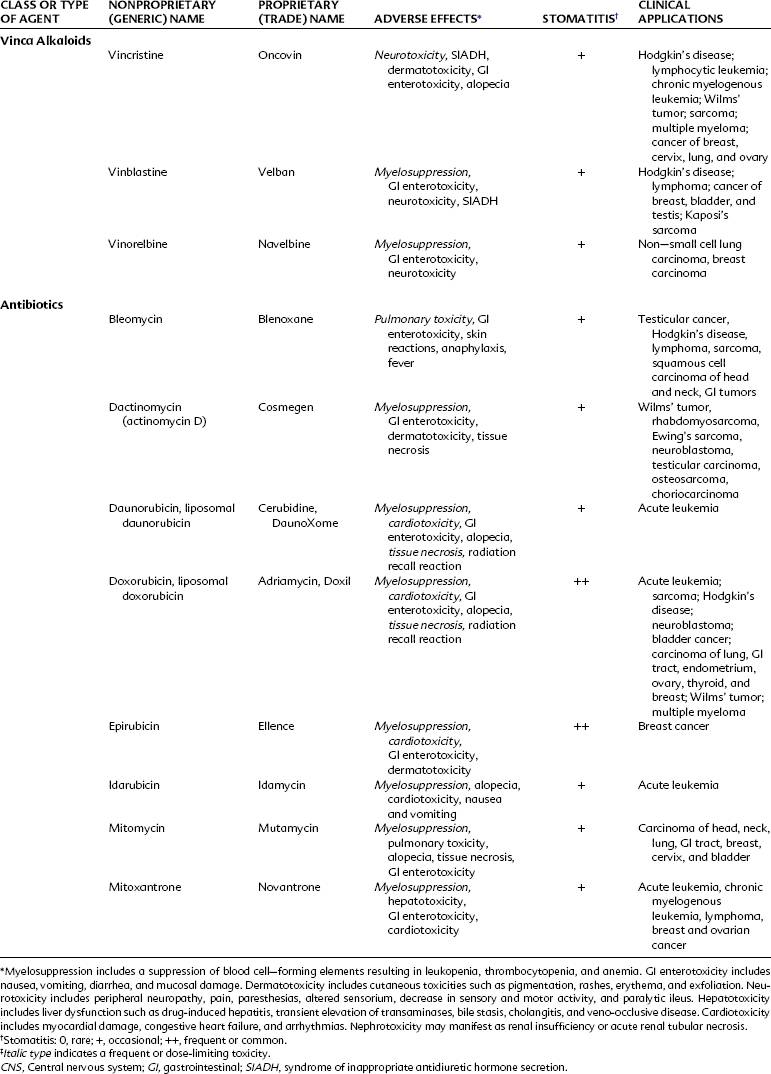
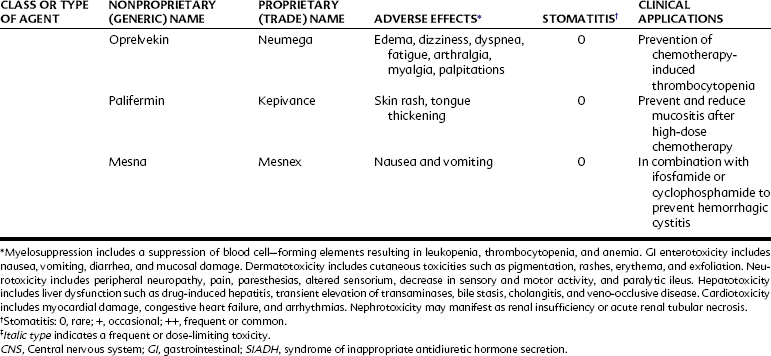
Alkylating agents are not cell cycle specific, although they are most destructive to rapidly proliferating tissues and seem to cause cellular death only when the cell attempts to divide. Because they produce irreversible changes in the DNA molecule, alkylating agents are mutagenic, teratogenic, and carcinogenic in addition to being oncolytic. Alkylating agents are also radiomimetic because they produce morphologic damage in cells similar to the damage caused by radiation injury. Because most of these agents are myelosuppressive, immunosuppression and susceptibility to infection are common outcomes. They vary greatly in lipid solubility, membrane transport, and pharmacokinetic properties and differ in clinical use. The molecular structures of representative alkylating agents are shown in Figure 42-3; adverse effects and clinical applications are summarized in Table 42-1.
Nitrogen mustards
Bendamustine hydrochloride
Bendamustine hydrochloride (Treanda) is an intravenously administered bifunctional mechlorethamine derivative with alkylator and purine antimetabolite activity. This bifunctional agent may have an advantage to overcome cross-resistance with other alkylating agents. Bendamustine has been studied in combination with rituximab in the management of patients with indolent or mantle cell lymphoma and has shown significant activity.59 It has been approved more recently for the treatment of chronic lymphocytic leukemia and continues to be studied in many other tumor types, including breast cancer and sarcomas. The most common adverse effects (occurring 15% to 20% of the time) include neutropenia, thrombocytopenia, anemia, pyrexia, nausea, and vomiting.
Tetrazines
Temozolomide
Temozolomide is the first imidazotetrazinone derivative used in clinical practice. Similar to DTIC, temozolomide is metabolized to monomethyl 5-triazinoimidazole carboxamide (MTIC), which is ultimately converted to the cytotoxic methyldiazonium ion. Temozolomide has advantages over DTIC: it can be administered orally, and it does not require hepatic conversion to MTIC because temozolomide is spontaneously converted to the active metabolite at physiologic pH.29 Temozolomide penetrates tissues well and is able to cross the blood-brain barrier, allowing it to be used to treat brain tumors such as astrocytoma69 and glioblastoma multiforme, an aggressive primary brain tumor. Temozolomide has also been used to treat malignant melanoma. The major toxic effects associated with this alkylating agent include myelosuppression, nausea, vomiting, headache, and fatigue.
Nitrosoureas
Streptozocin
Streptozocin is a naturally occurring antibiotic that has a mode of action similar to that of nitrosoureas. In contrast to carmustine and lomustine, however, streptozocin does not readily cross the blood-brain barrier, and it is not strongly myelosuppressive. Streptozocin is unique in its special affinity for the islet cells of the pancreas. The drug is diabetogenic in animals and effective against metastatic insulinomas in humans. Streptozocin should be administered intravenously with care because it is a vesicant. It is one of the most emetogenic agents and requires adequate premedication with antiemetics. Potentially fatal renal toxicity and hepatotoxicity have occurred.67
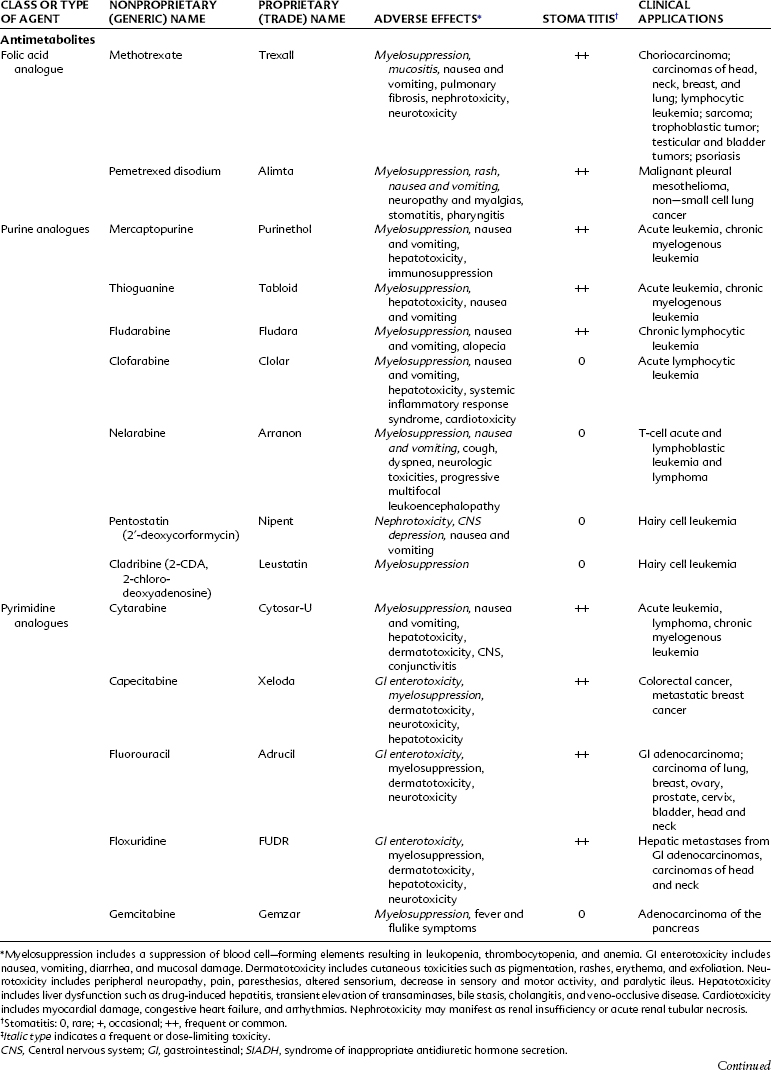
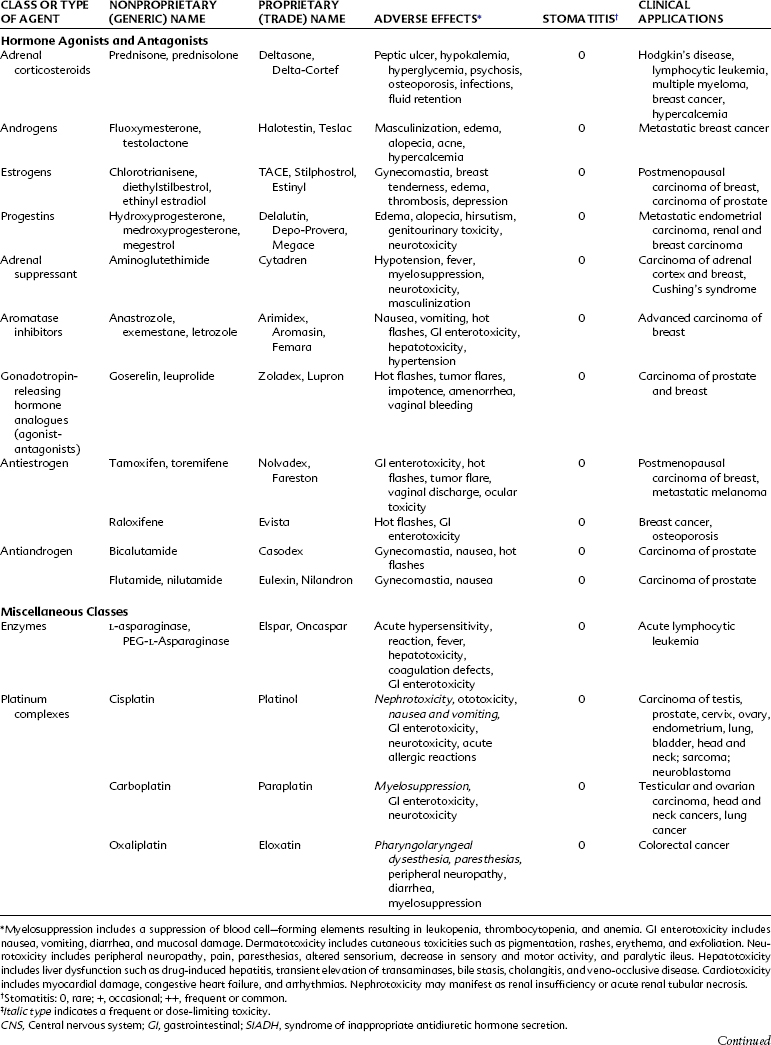
Antimetabolite Agents
Antimetabolites bear a marked structural resemblance to folic acid and to the purine and pyrimidine bases involved in the synthesis of DNA, RNA, and certain coenzymes (Figure 42-4). They differ in molecular arrangement from the corresponding metabolite to a degree sufficient to serve as fraudulent substrates for biochemical reactions, either inhibiting synthetic steps or becoming incorporated into molecules and interfering with cellular function or replication. Antimetabolites characteristically exert their major effects during the S (DNA synthesis) phase of the cell cycle. This activity interferes with the growth of rapidly proliferating cells throughout the body—the bone marrow, germinal cells, hair follicles, and lining of the alimentary tract. Oral manifestations are an especially prominent feature of the toxicity of these agents. Three classes of antimetabolites exist: folic acid analogues, purine analogues, and pyrimidine analogues.
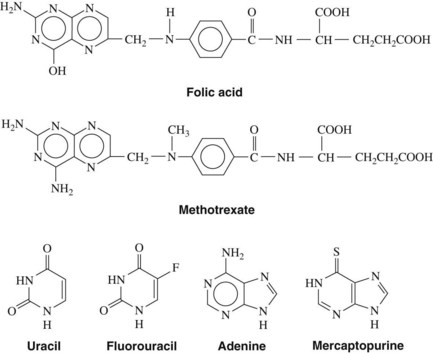
FIGURE 42-4 Structural relationships between several antimetabolites and their respective analogues.
Methotrexate
Methotrexate is subject to many important drug interactions. Highly plasma protein-bound drugs such as salicylates, sulfonamides, and phenytoin may displace methotrexate from its protein-binding sites and result in greater toxicity. Organic acids such as salicylate and probenecid inhibit the tubular secretion of methotrexate, resulting in increased concentrations of methotrexate and toxicity. Penicillins can also compete with methotrexate for renal tubular secretion.32 In patients receiving large gram doses of methotrexate, the concurrent use of nonsteroidal anti-inflammatory drugs (NSAIDs) should be avoided because this drug class can also reduce renal blood flow and increase the risk of nephrotoxicity.
Pemetrexed disodium
Pemetrexed disodium is a new antifolate that can attack multiple enzyme targets, including dihydrofolate reductase, thymidylate synthase, and glycinamide ribonucleotide formyl transferase. By inhibiting the formation of precursor purine and pyrimidine nucleotide, pemetrexed prevents the formation of DNA and RNA, which are required for the growth and survival of normal cells and cancer cells. Pemetrexed is approved for treatment of malignant pleural mesothelioma and for use as a second-line agent for treatment of non–small cell lung cancer (NSCLC). Vitamin supplementation with folic acid (1 mg daily) and vitamin B12 (1000 µg intramuscularly every 9 weeks) helps to control the hematologic and nonhematologic toxicities.49 Steroid treatment with dexamethasone (4 mg twice a day on the day before, the day of, and the day after pemetrexed therapy) is used to help limit skin rashes. The most common side effects are hematologic, rash, nausea, and vomiting. Occasionally, chest pain, edema, and hypertension may be seen in patients, and neuropathy and myalgias occur in 29% and 13% of patients, respectively. Stomatitis has been reported in about 20% of patients treated with pemetrexed.
Purine analogues
Mercaptopurine and thioguanine
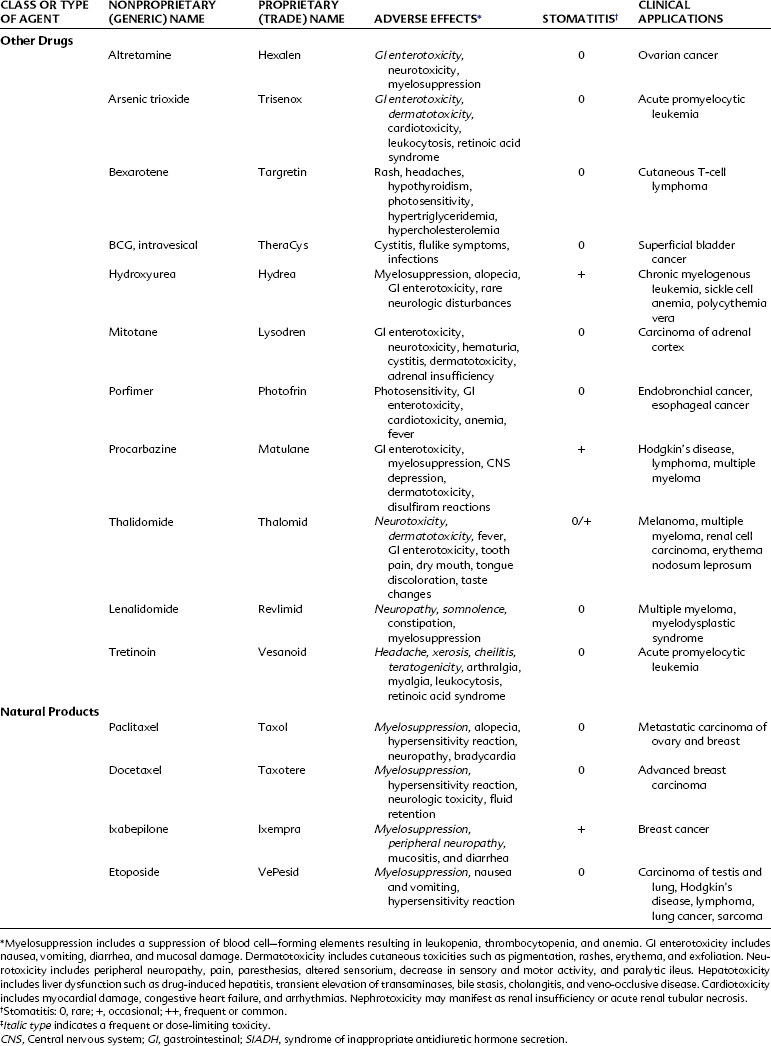
Orally administered mercaptopurine is readily absorbed but undergoes extensive first-pass metabolism by the liver. After intravenous injection, the plasma half-life is approximately 90 minutes. The drug is metabolized by methylation in the liver and by the hepatic enzyme xanthine oxidase. Concurrent administration with allopurinol, a xanthine oxidase inhibitor originally developed to increase the anticancer effect of mercaptopurine, requires a 50% reduction in the dose of mercaptopurine. Allopurinol is of little clinical value in this setting because it also increases the toxicity of mercaptopurine. The use of allopurinol in the treatment of gout is described in Chapter 21. Currently, mercaptopurine is used mainly for maintenance of remission in acute lymphocytic leukemia. The chief toxic effect is myelosuppression. Pulmonary fibrosis and pancreatitis may also occur. Thioguanine has activity, toxicity, and clinical applications similar to those of mercaptopurine.
Fludarabine
Fludarabine (2-fluoro-ara-AMP) is an analogue of adenosine. This injectable purine antagonist is quickly dephosphorylated in the plasma, enters the cell, and is converted to the triphosphate form. This false nucleotide inhibits ribonucleotide reductase and DNA polymerase, which results in the inhibition of DNA synthesis.37 Fludarabine is indicated for the treatment of B-cell chronic lymphocytic leukemia in patients who have not responded to traditional therapy with an alkylating agent. Fludarabine is primarily excreted by the kidneys and has a long plasma half-life of approximately 10 hours. Transient myelosuppression and immunosuppression, with an increased risk of opportunistic infection, seems to be the major toxicity at current doses. Fludarabine has also been used for treatment of non-Hodgkin’s lymphoma, hairy cell leukemia, and cutaneous T-cell lymphoma and in salvage regimens for the treatment of acute myeloid leukemia.
Clofarabine and nelarabine
Clofarabine and nelarabine are the newest purine nucleoside antimetabolites approved for the treatment of acute lymphocytic leukemias. Clofarabine is converted intracellularly by deoxycytidine kinase to the 5′-monophosphate metabolite, then via monophosphokinases and diphosphokinases to the active 5′-triphosphate form. The clofarabine 5′-triphosphate inhibits DNA synthesis through its action on ribonucleotide reductase and DNA polymerases. Clofarabine is approved by the U.S. Food and Drug Administration (FDA) for the treatment of pediatric patients with relapsed or refractory acute lymphocytic leukemia after at least two prior treatment regimens.24 It is also being studied for other malignancies, including the treatment of acute myeloid leukemias in adults. The principal toxicities associated with clofarabine are nausea, vomiting, hematologic toxicity, febrile neutropenia, hepatobiliary toxicity, infections, and renal toxicity. Clofarabine can also produce a syndrome manifested by the rapid development of tachypnea, tachycardia, hypotension, shock, and multiorgan failure called systemic inflammatory response syndrome, which is similar to a capillary leak syndrome. Cardiac effects include tachycardia and left ventricular systolic dysfunction.
Nelarabine is a prodrug of the deoxyguanosine analogue-9-beta-D-arabinofuranosylguanine (ara-G). Nelarabine is demethylated to ara-G and activated to the active 5′-triphosphate, ara-GTP. The active ara-G is incorporated into the DNA resulting in inhibition of DNA synthesis and cell death. There is a differential accumulation in T cells, and nelarabine is approved for the treatment of patients with T-cell acute lymphoblastic leukemia and lymphoma who have not responded to or have relapsed after treatment with at least two other chemotherapy regimens.9 A major adverse effect associated with nelarabine resulting in a “black box” warning involves neurologic events that include severe somnolence, convulsions, peripheral neuropathies, and paralysis. Other adverse effects include fatigue, bone marrow suppression, gastrointestinal side effects, and some pulmonary complaints of cough and dyspnea. Rarely, patients have complained of blurred vision while receiving nelarabine. The combination of nelarabine and adenosine deaminase inhibitors such as pentostatin should be avoided because this combination may result in a decreased conversion of nelarabine to its active substrate, decreasing its efficacy and potentially changing the adverse profile of both drugs.
Pyrimidine analogues
Fluorouracil and floxuridine
The fluorinated pyrimidines fluorouracil and floxuridine are prepared by substituting a stable fluorine atom for hydrogen in position 5 of the uracil and deoxyuridine molecules. These compounds, after intracellular conversion to 5-fluoro-2′-deoxyuridine monophosphate, are potent antimetabolites that bind to and inhibit thymidylate synthetase, inhibiting formation of thymidylic acid and impairing DNA synthesis. Fluorouracil metabolism also produces a critical intermediate, 5-fluorouridine triphosphate, which is incorporated into RNA and interferes with its function. 5-Fluorodeoxyuridine triphosphate (5-FdUTP) may also be incorporated into DNA, producing single-strand breaks contributing to the cytotoxicity.54
Capecitabine
Capecitabine (5′-deoxy-5-fluoro-N-[(pentuloxy)carbonyl]-cytidine) is a newer oral agent used in the treatment of advanced breast and colorectal cancers. Capecitabine is hydrolyzed in the liver and ultimately converted to the active drug 5-fluorouracil. Its activity profile and pharmacokinetic profile are similar to infusional fluorouracil. Side effects of capecitabine include severe diarrhea, stomatitis, and some mild nausea and vomiting. Severe hand-foot syndrome (palmar/plantar erythrodysesthesia) and other dermatologic changes have been reported.16
Cytarabine
Cytarabine (cytosine arabinoside) is an analogue of 2′-deoxycytidine that can inhibit DNA synthesis by inhibiting DNA polymerase activity as a result of its incorporation into DNA and the formation of fraudulent DNA. Premature DNA chain termination results. Cytarabine is primarily a cell cycle S phase–specific agent. When given intravenously, the drug is rapidly cleared from the blood by deamination in the liver, with a plasma half-life of 5 to 20 minutes. With these properties, continuous infusion is often the preferred route of administration. Cytarabine crosses the blood-brain barrier, achieving cerebrospinal fluid concentrations of 40% to 50% of those of plasma. This feature allows for the treatment of CNS disease with systemic high-dose therapy. Cytarabine may be administered intrathecally and produces high concentrations that decline slowly because of the absence of cytidine deaminase in the CNS.1 Cytarabine is the most active single drug available for the treatment of acute myelogenous leukemia in adults, producing about a 25% incidence of complete remission. It is often used in combination with other agents. It has some modest activity against lymphomas. The major side effect is myelosuppression. High doses produce severe nausea and vomiting, severe diarrhea, cerebellar toxicity, and keratoconjunctivitis.26
Gemcitabine
Gemcitabine (difluorodeoxycytidine) is a newer antimetabolite useful in many experimental tumor models, with clinical responses in NSCLC and breast cancer. It is currently indicated for first-line treatment of patients with locally advanced or metastatic adenocarcinoma of the pancreas.31 Recent trials support the use of gemcitabine in combination with cisplatin to treat metastatic NSCLC. Its dose-limiting side effect is myelosuppression characterized by thrombocytopenia. Transient febrile episodes and a flulike syndrome have been commonly reported.
Antibiotics
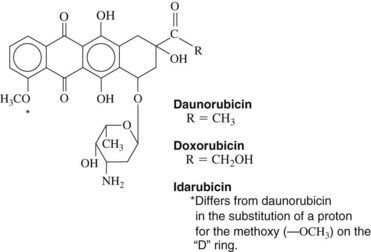
Daunorubicin
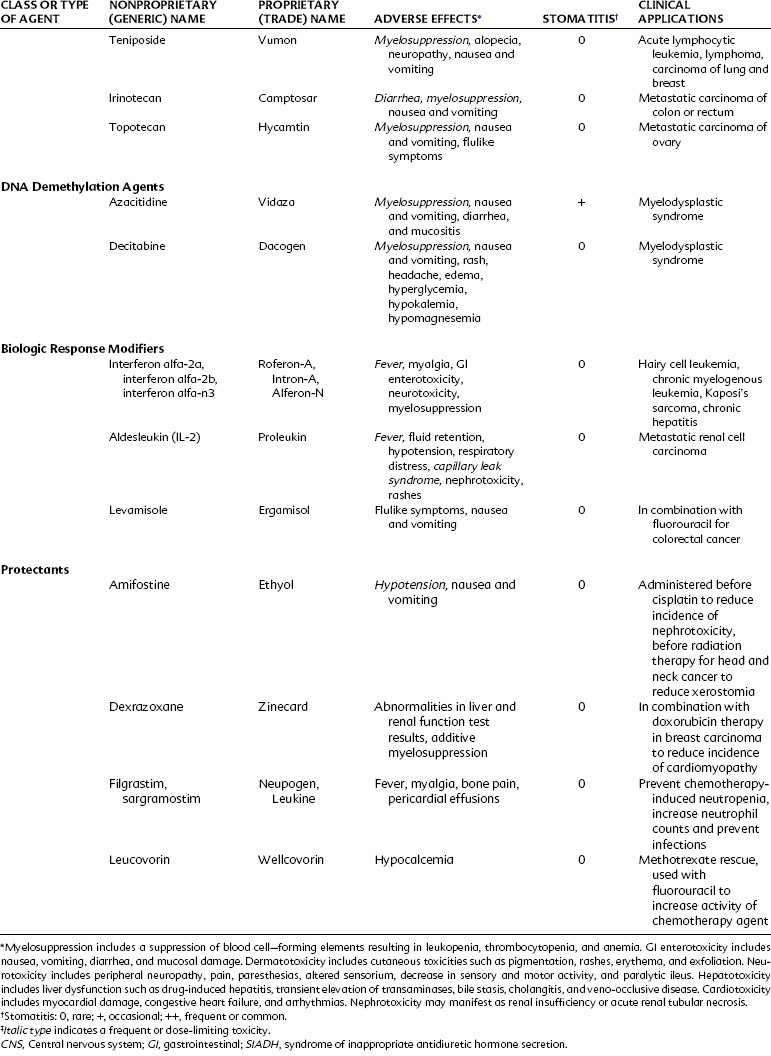
Daunorubicin is a cytotoxic anthracycline antibiotic produced by Streptomyces peucetius subsp. caesius, which is also the source of doxorubicin and idarubicin (Figure 42-5). The drug combines with DNA in an intercalative mode by slipping into the helical structure between stacked bases. Synthesis of DNA and RNA is inhibited, and preformed DNA is damaged. Other possible mechanisms are postulated, including metabolism to form cytotoxic free radicals, a cell membrane surface cytotoxic action, and inhibition of topoisomerase II. The killing effect of daunorubicin is at a maximum in the DNA synthesis S phase of the cell cycle, but damage is not phase specific. Experimental evidence exists for synergy between these antibiotics and drugs such as etoposide.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


