CHAPTER 38 Principles of Antibiotic Therapy*
INFECTIOUS DISEASE PAST AND PRESENT
In 1967 the U.S. Surgeon General declared, “The time has come to close the book on infectious diseases.” In 1993, 17 million people died of infectious diseases throughout the world, with 11.4 million deaths (mostly of children) caused by bacterial diarrhea and pneumonia. In the same year, 15.6 million died of cardiovascular disease and cancer combined.69 The four primary disease killers caused by infection are the same as in 1900: diarrhea, pneumonia, tuberculosis, and malaria.19 One third of the world population has tuberculosis, and Africa accounts for 90% of the 300 to 500 million new cases of malaria annually, with 1.5 to 2.7 million deaths per year. In World War II, 55 million people were killed; by 2010, 65 million will have died of acquired immunodeficiency syndrome (AIDS).
The U.S. Centers for Disease Control and Prevention estimates that 65,000 to 90,000 deaths annually in U.S. hospitals result from nosocomial (hospital-acquired) infections. This figure may be a significant underestimate and the number may be closer to 200,000 to 300,000 because infectious disease deaths may be misclassified as cardiac arrest or respiratory or renal failure instead of their underlying microbial causes. In 1977, 100,000 gram-negative nosocomial bacteremic deaths were estimated annually in the United States57; bloodstream infections (septicemia and bacteremia) alone, among all nosocomial infections, may now be the eighth leading cause of death in the United States.114,115
The importance of two medical discoveries that have essentially doubled the human life span in first-world countries since the 1850s—anesthesia and the control of infectious diseases—cannot be overestimated. Without the ability to operate internally within the human body free of excruciating pain, the gains of medical and dental surgery would be void. In the United States in 1776, the average life span was less than 40 years of age. In England in 1853, infectious disease was responsible for 37% of all deaths. At the beginning of the twentieth century in the United States, the infant mortality rate was 100 per 1000; now it is less that 10 per 1000.69 A child in 1900 had a 10% chance of death between ages 1 and 4 years from pneumonia or diarrhea.69 Many adults, infants, and children died of typhus, typhoid, diphtheria, whooping cough, yellow fever, malaria, influenza, measles, smallpox, and streptococcal and staphylococcal infections.
Even before the advent of the modern germ theory of disease in the 1870s, many individuals surmised that filth had a substantial bearing on disease. The “sanitary movement” began in Great Britain in the 1850s and the United States in the 1870s with improvement in wages, housing, education, and personal hygiene. Civil engineers cleaned the streets, water, and air, and cities removed refuse and their attendant rodent vectors of disease. Waste disposal, clean water, and hand hygiene by public health engineering have reduced the transmission of 35 to 40 infectious diseases.31
The modern era of infectious disease began with the first visualization of microbes by Anton van Leeuwenhoek in 1683, the “animicules” of dental plaque scraped from his upper gingiva and killed with salt (the first periodontal chemotherapy).47 In 1776, Edward Jenner administered the first smallpox vaccination. In 1848, Ignaz Semmelweiss introduced clean surgical operating technique (“gentlemen, wash your hands”). In 1854, John Snow showed the link between cholera and drinking water.47
In the 1860s, Louis Pasteur first used the word germ for living entities that produced disease, and Joseph Lister used carbolic acid to disinfect wounds. In the 1870s, Robert Koch proved the bacterial causation of anthrax and tuberculosis, and in the 1880s, Pasteur developed anthrax and rabies vaccines. In 1891, Paul Ehrlich showed that antibodies were responsible for immunity. In 1897, Ivanowski and Beiternick discovered viruses. The mosquito vector for yellow fever was shown in 1900, Treponema pallidum was found to be the cause of syphilis in 1905, human immunodeficiency virus (HIV) was identified in 1983, Helicobacter pylori was discovered as a cause of peptic ulcer in 1984, and the West Nile virus was identified in 1999.47
In the early 1900s, Paul Ehrlich used the term magic bullet for his predicted chemical that would affect only microbial cells and have no effect on mammalian cells. He later used fuchsin and mercury (Salvarsan) to treat syphilis. In 1928, Alexander Fleming serendipitously discovered that a mold, Penicillium chrysogenum, lysed staphylococci; this was later developed to its full potential by the isolation of penicillin from Penicillium notatum by Florey and colleagues at Oxford in the late 1930s and early 1940s. The first use of penicillin was in 1941 on an English police constable with streptococcal and staphylococcal skin abscesses. In the United States, penicillin was first used in 1942 on Anne Miller, who had streptococcal toxemia of pregnancy. All of these firsts have possibly overshadowed arguably the greatest of all medical advances: the demonstration in 1935 by Gerhard Domagk that sulfanilamide could be safely used systemically to treat infectious disease. The “dreaded disease of summer” (poliomyelitis) declined from 57,879 cases in the United States in 1953 to 72 cases in 1965 with the advent of the polio vaccine.71 By 1977, smallpox was eradicated from the world as a contagious disease. During 1900-1997, the American life span increased by 60% to a median age of 76.19
In the developing world, a different story has unfolded. In 1998, the World Health Organization determined that infectious disease caused 25% (13 million) of the 54 million deaths in the world that year, with pneumonia (3.5 million), AIDS (2.3 million), diarrhea (2.2 million), tuberculosis (1.5 million), malaria (1.1 million), and measles (1 million) the top killers.19 The incidence of emerging infections (defined by the Institute of Medicine as new, re-emerging, or drug-resistant infections whose incidence has increased in the last 2 decades or whose incidence threatens to increase) has increased.19 Now included in this category are legionnaires disease, toxic shock syndrome, respiratory syncytial virus, Lyme disease, Nipah virus, Hantavirus, hemorrhagic viral diseases (dengue, Ebola, Marburg), Escherichia coli O157:H7, malaria, yellow fever, cholera, and multidrug-resistant tuberculosis. All of these infections and more are potentially transmitted by 500,000 world refugees and 1.6 billion annual airline passengers, 500 million of which cross borders each year.69
All of the media attention to these potential pathogens has led to a second “germ panic” with the revival of the focal infection theory of disease,73 which alleges that many or most current diseases are caused by various microbes, including cardiovascular disease; various forms of emotional disorders such as obsessive-compulsive disorder, Tourette’s syndrome, autism, and schizophrenia; preterm births; chronic fatigue syndrome; and multiple sclerosis. The first germ panic of 1900-1940 was fostered by the focal infection theory as espoused by Hunter and colleagues, in which a localized infection in one area of the body could move and occur elsewhere in the body and cause various pathologic conditions, such as arthritis, neuritis, myalgia, osteomyelitis, endocarditis, brain abscess, skin abscess, pneumonia, anemia, indigestion, gastritis, pancreatitis, colitis, diabetes, emphysema, goiter, thyroiditis, Hodgkin’s disease, “obscure fever,” nervous diseases, headache, mental apathy, and mental incompetence.73 All of these were disorders for which medicine at the time (and many currently) had no explanations and no answers.
These foci of infection were conveniently located in areas of the body readily accessible to surgery (particularly in the wealthy): teeth, tonsils, and the facial sinuses, leading to an excessive number of dental extractions, tonsillectomies, and other surgeries in the first half of the twentieth century.42,73,117,118 The resurrection of the foci of infection concept today is based on limited scientific evidence and questionable studies that lack attention to sound epidemiologic methods.
Very rarely, microbes leave the oral cavity and metastasize to other areas of the body to initiate a nonspecific inflammatory infectious process manifested as liver, splenic, or brain abscesses or bacterial endocarditis. These microorganisms are almost always VGS and almost never pathogens associated with periodontal disease. That these metastatic infections are so rare is truly remarkable and speaks well for our immune defense mechanisms, particularly in the oral cavity and blood, and the reticence of microorganisms to leave their ecologic niches for foreign environments. Currently, little evidence suggests that the oral cavity is the source of significant systemic disease.72,73
PATTERNS OF ANTIBIOTIC USE AND MISUSE
Antibiotics are the most widely abused prescribed drugs on the basis of inappropriate indications, dosages, and duration of use. Approximately half of all antibiotics used in hospitals are given to patients without signs or symptoms of infection, in many cases to “prevent” infections and to ensure that “everything was done” to avoid later criticism. Antibiotics are often used as “drugs of fear”45 to cover for potential errors of omission or commission and prevent a claim of negligence. The abuse of negligence (tort) law has been a major contributing factor to the massive overuse of antibiotics and the attendant mortality rate associated with highly antibiotic-resistant microorganisms.
In hospitals, one third of antibiotics are used empirically, one third for prophylaxis, and one third with appropriate culture and sensitivity tests.69 Because hospitals save money by not using culture and sensitivity tests, the demand has been for broader spectrum antibiotics, which has created a vicious cycle by disturbing the hospital microbial ecology further and fostering even greater microbial resistance.98
Outpatient antibiotic use is characterized by the “80 : 80 rule”: 80% of all antibiotics are used in the community, and 80% are used for respiratory infections—most of which are viral in cause and not amenable to antibiotic therapy.69 Of the 50% of people with acute respiratory illness who seek medical treatment, 50% to 80% may receive an antibiotic, but pneumonia (the only respiratory tract disorder requiring an antibiotic) may account for only 2% of these cases.
The prescribing of antibiotics can vary 15-fold among physicians. Physicians who tend to prescribe many drugs also prescribe many antibiotics. Antibiotic prescriptions are a quick way to end an office visit and reduce return visits.90
Dentists prescribe 7% to 11% of all common antibiotics (β-lactams, macrolides, tetracycline, metronidazole, clindamycin), and abuse of such antibiotics can be substantial.18 In England, 33% to 87% of various antibiotics were judged to be inappropriately prescribed by dentists according to the Dental Practitioners Formulary.74 Experts in England are in agreement that antibiotics are used too long for the management of orofacial infections and that shorter durations are more appropriate and reduce the selection of drug-resistant microbes.56
In a survey of 505 Canadian dentists, the average length of antibiotic therapy was 6.92 days (range 1 to 21 days), and 17.5% did not use the 1997 American Heart Association (AHA) guidelines for endocarditis prophylaxis.27 Two thirds of the dentists used antibiotic prophylaxis for patients with rheumatic fever without rheumatic heart disease; 25%, for patients with HIV/AIDS; 70%, for prosthetic joints; and two thirds, for restorative dentistry not associated with significant bleeding even though not advocated by the AHA.
AHA prophylaxis for patients with cardiac valve prostheses was not used by 20% of dental specialists. The study concluded that antibiotics are underused for symptomatic infections, overused for surgical prophylaxis, and commonly used at suboptimal dosing with prolonged dosing schedules and often not according to antibiotic prophylaxis guidelines.27
In a survey of antibiotic use by 1606 members of the American Association of Endodontists, 12.5% used antibiotics as an analgesic for post-treatment pain; 37.3%, as antibiotic prophylaxis after surgery; 44.8%, after incision and drainage without systemic involvement or patient immunosuppression; and 12% to 54%, for situations in which they are not effective, such as the following: (1) irreversible pulpitis with moderate-severe symptoms with or without apical periodontitis; (2) asymptomatic necrotic pulps with chronic apical periodontitis but no swelling; (3) necrotic pulps with acute apical periodontitis, no swelling, and moderate-severe symptoms; and (4) asymptomatic necrotic pulps with chronic periapical periodontitis with or without a sinus tract.119 The authors concluded that not much had changed in the past 25 years.
Inappropriate antibiotic use in dentistry includes the following situations: (1) antibiotic therapy initiated after surgery to prevent an infection unlikely to occur and not documented effective for this purpose by clinical trials; (2) failure to use prophylactic antibiotics according to the principles established for such use; (3) use of antibiotics as analgesics in endodontics; (4) overuse in situations in which patients are not at risk for metastatic infections; (5) treatment of chronic periodontitis almost totally amenable to mechanical therapy; (6) administration of antibiotics instead of mechanical therapy for periodontitis; (7) long-term administration in the management of periodontal diseases; (8) antibiotic therapy instead of incision and drainage; (9) administration of antibiotics to avoid claims of negligence; and (10) administration in improper situations, dosage, and duration of therapy.69
ANTIBIOTIC MECHANISMS OF ACTION
Antimicrobials affect the viability of microorganisms by five known processes: (1) inhibition of cell wall synthesis, (2) alteration of cell membrane integrity, (3) inhibition of ribosomal protein synthesis, (4) suppression of deoxyribonucleic acid (DNA) synthesis, and (5) inhibition of folic acid synthesis (Table 38-1, Figure 38-1). Microbial cell wall synthesis inhibition and membrane effects are extracytoplasmic, and inhibition of nucleic acid, protein, and folic acid synthesis is intracytoplasmic. Drugs that affect bacterial cell wall or membrane integrity and DNA synthesis are usually, but not always, bactericidal (inducing cell death), and protein and folic acid synthesis inhibitors are usually bacteriostatic (preventing cell growth or replication).
TABLE 38-1 Mechanisms of Action of Common Antibiotics

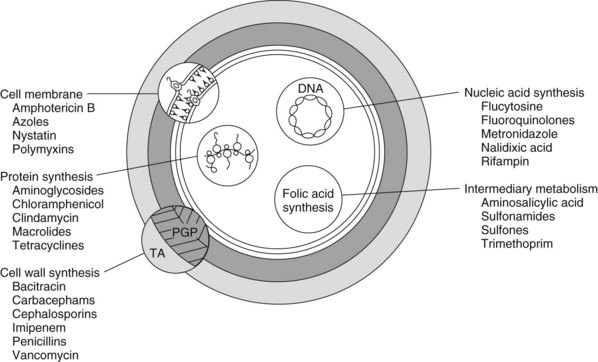
FIGURE 38-1 Site and mechanism of action of antimicrobial agents. PGP, Peptidoglycan; TA, teichoic acid.
MICROBIAL RESISTANCE TO ANTIBIOTICS
All microbial resistance is local; the patterns and extent of this resistance are determined by the use of antibiotics in a particular community. What is true in Florida may not be true in Los Angeles or in Paris, London, Rome, or New Delhi. If tetracyclines are used widely in the community for acne or Lyme disease, a high resistance level to the drug is likely to be present in that locale. If not, the microbial resistance level is likely to be low. If an antibiotic or its analogue has been used widely in agriculture, this may strongly influence resistance patterns—to the point of rendering a new antibiotic far less useful. In Taiwan, virginiamycin (a streptogramin) has been used for more than 2 decades as a growth promoter in food animals. When quinupristin-dalfopristin, a new streptogramin, was tested on human bacterial isolates before its clinical introduction, more than 50% of some pathogens were already resistant to the drug. Antibiotics are truly societal drugs that cumulatively affect the individual receiving the drug and many others as well.50
Microorganisms have developed seven known mechanisms to evade the bactericidal or bacteriostatic actions of antimicrobials, as follows: (1) enzymatic inactivation, (2) modification/protection of the target site, (3) limited access of antibiotic (altered cell membrane permeability), (4) active drug efflux, (5) failure to activate the antibiotic, (6) use of alternative growth requirements, and (7) overproduction of target sites (Table 38-2).69,88
TABLE 38-2 Antibiotic Resistance Mechanisms

Most experts agree that the major factor in the development and maintenance of antibiotic resistance in microbes is their ability to eliminate sensitive microorganisms and allow resistant ones to multiply and dominate. Although this selection process is crucial, other factors also contribute. Lengthy antibiotic regimens are commonly advocated to kill all the resistant strains or prevent stepped resistance (the development of resistance by a sequence of mutations occurring over several generations of microbial multiplication). Theoretically, if the antibiotic is given long enough, all these mutants are exposed to the antibiotic and killed at cell division. This is the rationale for taking the entire prescribed antibiotic rather than stopping the antibiotic when the patient is well. This concept is false for three reasons: (1) microbial mutations rarely occur during antibiotic treatment; (2) stepped resistance occurs even with prolonged antibiotic use4; and (3) most antibiotic resistance is gained by the transfer of genetic material between microbes, which is greatly enhanced by low-dose, prolonged antibiotic therapy.50 Combination antibiotic therapy against the stepped resistance seen with Mycobacterium tuberculosis is unique for this organism, but should not be extrapolated to all microbes. Also, a directive to “take all the antibiotic” assumes that the prescriber knows the exact duration of the infection, which is impossible.
Microbial resistance is most likely to occur when subtherapeutic antibiotic doses are used—doses that do not kill or inhibit the microorganism, but rather allow it to perceive the chemical as a threat to its survival and to react by mutation to resistance, acquisition, or transfer of resistance genes/virulence factors or induction (expression) of latent resistance genes.32,50 The gastrointestinal tract is a massive reservoir for resistance genes readily transferred within and between enteric microbial species,93 a process greatly enhanced by antibiotics that readily induce the expression or transfer of resistance genes, such as tetracyclines, imipenem, cefoxitin, and clavulanic acid.82
SPECIFIC RESISTANCE MECHANISMS
β-Lactamases
The most important acquired mechanism for β-lactam resistance, particularly in gram-negative microorganisms, is the production of various β-lactamases that hydrolyze the β-lactam ring to form a linear metabolite incapable of binding to PBPs. In 1984, 19 plasmid-mediated β-lactamases were known; the number now has increased to more than 340 chromosomally and plasmid-mediated β-lactamases—70 of the TEM-1 and TEM-2 and 20 of the SHV-1 types alone.11
β-Lactamases have been variously classified by Richmond-Sykes (I to V), Ambler (A to D), and Bush (1 to 4).11 β-Lactamase enzymes can be chromosomally mediated or easily transferred by transposable elements. Many are of the TEM type (from a patient named Temoniera in Greece, in whom a β-lactamase was isolated in the early 1960s) or the SHV type (sulfhydryl variable).11 The most pressing difficulties with β-lactamases are their widespread dissemination throughout the microbial environment, ability to move between widely disparate organisms, tendency to inhibit new antibiotic agents rapidly, and increasing resistance to β-lactamase inhibitors (clavulanic acid, sulbactam, and tazobactam). β-Lactamases have been observed in numerous gram-positive and gram-negative pathogens. The cohabitation of staphylococci and enterococci on human skin in hospitals has likely led to the incorporation of β-lactamase genes into enterococci after the latter organisms had successfully avoided this transfer for billions of years.
The first plasmid encoded β-lactamases with the ability to hydrolyze cephalosporins were termed the extended-spectrum β-lactamases (ESBLs).13 These ESBLs microbes were also resistant to aminoglycosides, tetracyclines, and trimethoprim/sulfonamides.13 ESBLs cause resistance to third-generation cephalosporins (cefotaxime, ceftriaxone, ceftazidime) and monobactams (aztreonam) but are sensitive to cefamycins (cefoxitin, cefotetan) and carbapenems (imipenem, meropenem, ertapenem). These ESBLs are horizontally transmitted by mobile genetic elements from food, animals, or family members and induce greater mortality than enteric bacilli without these ESBLs.77
Multidrug Antibiotic Efflux Pumps
A mechanism by which bacteria move an antibiotic out of the cell as soon as it enters was first detected in E. coli by Levy in 1978; the first gene (qacA) encoding a multidrug efflux protein was subsequently detected in an isolate of S. aureus.69 Currently, more than 50 such systems have been described, and these cytoplasmic membrane transport proteins (multidrug efflux pumps) have likely evolved to protect the cell from foreign chemical invasion and allow its secretion of cell metabolic products.110 Efflux pumps operate in E. coli, Pseudomonas aeruginosa, staphylococci, Streptococcus pyogenes and S. pneumoniae, Bacillus subtilis, Pasteurella multocida, Neisseria gonorrhoeae, mycobacteria, and enterococci.110 For tetracyclines, these efflux pumps are the major mechanism for resistance and are becoming increasingly so for the fluoroquinolones.110 Efflux pumps are classified into five main groups: (1) the major facilitator family; (2) the small/staphylococcal multidrug resistance family; (3) the resistance, nodulation, and cell division family; (4) the adenosine 5′-triphosphate binding cassette superfamily; and (5) the multidrug and toxic compound extrusion family.110
These chromosomal and plasmid-mediated efflux transporter proteins may be quite specific for antibiotics and metabolic product substrates and are regulated by numerous genes and gene products. Repressors are also present and are highly regulated to prevent the accidental overproduction of efflux pumps. Tetracyclines derepress this system, leading to an overproduction of efflux proteins and increasing resistance to themselves and any other antibiotics carried by these proteins.81
Transposable Elements
Transposons are DNA segments that cannot self-replicate, but can self-transfer between plasmids, bacteriophages, and chromosomes. Transposons can recruit as many genes as required for their purpose, and the mechanisms that control this process are essentially unknown. That we know so little about a system with so much potential for genetic change is worrisome. Between 30% and 40% of the human genome is composed of transposable element sequences or gene sequences directly derived from them.23
Researchers previously hoped that resistance genes and their transporters would pose such a fitness problem for bacteria (requiring so much energy) that bacteria no longer exposed to the antibiotic would lose their resistance genes. Such genes may become so important for bacterial functions, however, that they become permanent. The tetracycline efflux pumps can become necessary for bacterial survival by functioning in Na+-K+ exchange across the bacterial membrane.6 The problem is compounded when the resistance gene for a particular antibiotic becomes part of an integron-containing multiple antibiotic resistance gene array. Eliminating the one antibiotic does nothing; all the antibiotics must be eliminated from the environment for the integron to be lost.
Integrons
Antibiotic resistance has been enhanced further by the discovery of the integron, a genetic element that captures and disseminates genes by site-specific integration of DNA (gene cassettes) that can mediate resistance, virulence, and biochemical functions.79 Integrons have three distinct genes encoding for an integrase enzyme, a recombination site, and a promoter element.79 Integrons resemble bundled products with a computer operating system; they package resistance determinants to allow for widespread gene dissemination.
Each gene is a cassette, and five genes may commonly be present in one integron.79 Superintegrons have been isolated in Vibrio cholerae that contain hundreds of gene cassettes that encode many bacterial functions beyond those of resistance and virulence. Gene cassettes have been identified for all antibiotics except fluoroquinolones, and they exist for quaternary ammonium compounds. Integrons cannot promote self-transfer because they lack transporter genes, but they are commonly associated with transposons and conjugative plasmids.
Horizontal Gene Transfer
Horizontal gene transfer (HGT), also known as lateral gene transfer, has been a major impetus for the exceptional diversity and survival of the microbial world. Ancient integrons, bacteriophages, plasmids, transposons, and now insertion sequence common regions have changed the otherwise clonal mode of prokaryotic life95 and allowed for gene capture and dissemination from the global gene pool.8 These mobile bacterial elements move from one bacterial cell to another via transduction and conjugation as circular, usually double-stranded DNA and often via a sex pilus as an extension from the donor to the recipient cell. Technically, a third form of DNA transfer is via transformation whereby “naked” DNA from disrupted cells is taken up from the extracellular fluid by certain “competent” cells.
These mobile genetic elements include plasmids, transopsons, bacteriophages, integrons, and insertion sequence common regions that move from bacterial cell to bacterial cell or, when translocated, from various DNA sites within the bacterial cell.8 These DNA elements carry genes for antibiotic resistance, heavy metal toxins, and virulence determinants and can induce or repair DNA damage.8
HGT encompasses two processes that move genetic material from one bacterium to another to enable unique phenotypic characteristics or to translocate genes from one location (plasmids) to another (chromosomes).59 The combination of integrons, transposons, and insertion elements results in antimicrobial resistance islands.59 Insertion sequences are mobile genetic elements that promote and translocate genes and are inserted into transposons to mobilize DNA resistance genes for β-lactamase of the CTX-M type.59
HGT facilitates genome rearrangement, deletions, and insertions to allow adaptation to changing environments and is induced by antimicrobials, metals, and organic contaminants (environmental stress).7 This facilitation of gene transfer occurs in four steps: (1) packaging of nucleic acids for transfer via excision and circularization of the transposons, (2) transfer of the DNA via contact with the recipient cell (conjugation), (3) entrance into the cell and integration with host chromosomal DNA, and (4) transfer of chromosomal DNA or the replicating elements into the daughter cells and subsequent generations.7
RESISTANCE IN MAJOR MICROBIAL PATHOGENS
Streptococcus pneumoniae
Microbial resistance to antibiotics in S. pneumoniae is most serious because the organism is responsible for 3000 cases of meningitis, 50,000 cases of bacteremia, 500,000 cases of pneumonia, and 2 million cases of otitis media in the United States annually and 3 to 5 million deaths annually worldwide.69 Resistance to sulfonamides was first detected in 1943 and to penicillin in the late 1960s in Australia and New Guinea. The mechanism of penicillin resistance is a single point mutation in PBP2x or PBP2b, with an altered PBP2a requiring mutation also in pBP2x (the organism has six PBPs).
High penicillin resistance (usually a plasma concentration of ≥2 µg/mL) is seen in 14% of U.S., 6.8% of Canadian, 10.4% of European, and 17.8% of Asian-Pacific isolates.38 Resistance of the pneumococcus to penicillin can vary significantly with geographic area: 38.8% in Tennessee, 15.3% in Maryland, 65.3% in Japan, 60.8% in Vietnam, 15.6% to 48.2% in Latin America, and 79.7% in Korea.69
Methicillin-Resistant Staphylococci
In a 1999 review of more than 10,000 bloodstream infections at 49 U.S. hospitals, S. aureus accounted for 16% and CoNS for 32% of all isolates, with most of the CoNS being methicillin-resistant.69 Some isolates are susceptible only to vancomycin; others are susceptible to linezolid and quinupristin-dalfopristin; and still others are susceptible to older agents such as macrolides, tetracyclines, aminoglycosides, rifampin, clindamycin, sulfonamides, and fluoroquinolones. The mechanism of methicillin resistance is an altered PBP2 (PBP2a or PBP2′) conferred through a mecA gene that results in a much lower binding affinity of methicillin for PBP2a. This mode of resistance requires the cooperation of PBP2 and PBP2a sites and two enzymes, one natural and one environmentally acquired.
Enterococci
Of the 17 species of enterococci found in the human oral cavity and gastrointestinal and genitourinary tracts, Enterococcus faecalis accounts for 90% of human infections, and E. faecium accounts for approximately 10%.69 Enterococcal infections are classic examples of a relatively harmless commensal organism becoming a serious pathogen by the acquisition of multiple resistance genes.
Enterococci are intrinsically resistant to cephalosporins and have varying degrees of resistance to aminoglycosides, macrolides, tetracyclines, and clindamycin. Vancomycin resistance, particularly in E. faecium, has been of major concern since the late 1980s. Enterococci cause 800,000 nosocomial infections annually in the United States, with more than 50% caused by vancomycin-resistant E. faecium; resistance is more than 90% in E. faecium bacteremias. Currently, 17% of enterococcal strains in the United States are resistant to vancomycin.52
Vancomycin-resistant enterococcus (VRE) infections, particularly of the bloodstream type, are becoming extremely difficult to treat. Doxycycline has been enlisted more recently in the treatment of VRE.52 Enterococcal resistance is complicated further by the observations that (1) streptococci, staphylococci, and enterococci often share the same resistance genes; (2) β-lactamase in enterococci is identical to that in staphylococci, indicating sharing of genetic information; (3) enterococci can transfer resistance genes, particularly for vancomycin to staphylococci and other organisms, in vitro and in animal models; (4) staphylococci and enterococci coinhabit the skin; and (5) the possibility exists that vancomycin resistance may one day appear in many VGS.69
Helicobacter pylori
Tetracycline has been added more recently to antibiotic regimens, and resistance rates are 0% in The Netherlands, 5.3% in Korea, and 58.8% in Shanghai, China, with the disturbing possibility that tetracycline-resistant H. pylori may exhibit cross-resistance with metronidazole. Metronidazole resistance in H. pylori may decrease the effectiveness of therapy by 37.7%, and clarithromycin resistance may decrease the effectiveness of therapy by 55%.25 The widespread use of systemic metronidazole and tetracycline is difficult to justify in the management of a relatively trivial, mechanically responsive disease such as periodontitis when such a practice may promote resistance in a microbial pathogen responsible for very serious diseases such as peptic ulcer and gastric cancer.
Human Immunodeficiency Virus
The current therapy for HIV infection entails highly active antiretroviral therapy (HAART) (see Chapter 40) with a combination of drugs that interfere with several steps in viral replication, including reverse transcriptase inhibitors, protease inhibitors, and the new integrase inhibitors that prevent HIV from integrating into the genome of the host cell. Difficulties have arisen with this therapeutic approach because the virus provides for reservoirs of replication-competent HIV in resting CD4 T lymphocytes throughout many years of intensive HAART. It is estimated that more than 60 years of HAART may be necessary to eradicate the virus from these reservoirs.69 More than 50% of HIV-infected individuals in the United States receiving HAART are resistant to one or more of the drugs, and 78% of individuals with measurable viral loads are resistant to at least one drug, encompassing about 100,000 people in the United States.99 From 1994-2000, 14% of new HIV cases had one or more HIV mutations associated with antiretroviral drug resistance; in 2000, it was 27%. Approximately 25% of newly infected, therapy-naïve individuals carry at least one key HIV drug-resistant mutant.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


