Chapter 3
Oral Carcinogenesis
It is estimated that approximately one in three people will develop cancer at some point in their life. Cancer affects almost every complex multicellular organism and involves disruption of the processes of cell proliferation, differentiation and development. Understanding cancer is not only important in the fight against malignant disease, but also in helping to improve our understanding of the fundamental biological mechanisms of life itself.
This chapter is concerned with the complex, multistage process of oral carcinogenesis. This is a cumulative sequence of cellular and tissue changes, some of which may be reversible but which if unchecked will ultimately transform normal oral keratinocytes into invasive cancer cells. It is important for us, as clinicians, to understand current pertinent concepts of oral oncology in order to recognise the earliest possible signs of carcinogenesis in our patients and to intervene proactively in the disease process.
Whilst the term oncology literally means the scientific study of new growths, it has become synonymous over the years with the overall study of malignant disease, the mechanisms governing cancer development, its treatment and clinical outcome. Traditionally, a malignant tumour is defined as an abnormal, uncoordinated growth of tissue that persists in an excessive manner after initiating stimuli have ceased and which then progresses to local tissue invasion, destruction and ultimately metastatic spread. In reality, malignant tumours are most likely composed of significantly heterogeneous and dynamic populations of abnormal and mutated cells that undergo constant and rapid cell proliferation and that compete for nutritional resource from both within the tumour and the outside host.
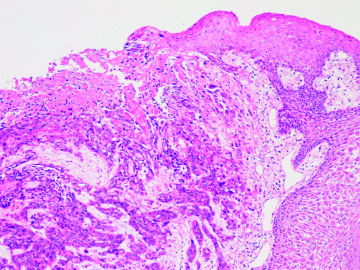
Figure 3.1 shows the histopathological appearance of an oral squamous cell carcinoma, which has arisen from a precursor dysplastic lesion, and which is now invading the underlying connective tissue. The overlying epithelium is no longer intact giving rise to the classic clinical picture of a non-healing, persistent mucosal ulcer.
Figure 3.1 Histopathology slide showing an oral squamous cell carcinoma arising from a pre-existing dysplastic lesion. On the surface there is a breach in continuity of the epithelium (ulceration), whilst carcinoma invades the immediately subjacent lamina propria.
Cancers may be initiated by various stimuli such as physical, chemical or biological agents and then, often following a latent phase, may be promoted by subsequent non-specific irritation or damage which encourages tumour progression and ultimately metastasis. Box 3.1 summarises these classic stages of initiation, promotion, progression and metastasis, which together form the hallmark of carcinogenesis.
Box 3.1 Classic stages in the development of cancer.
| • Initiation: | This is an irreversible process of DNA mutation, probably in stem cells, which follows damage induced by carcinogens. Mutagens may be physical, chemical or biological in nature |
| • Promotion: | Following a variable latent phase and, only after initiation, non-specific circumstances selectively ‘promote’ the expansion of altered cells, but are ineffective in producing cancer on their own. Cycles of initiation and promotion may exist before final emergence of the malignant phenotype |
| • Progression: | Increasing tissue disorganisation leads to a preinvasive state before finally progressing to malignant transformation, tissue invasion and local spread of disease |
| • Metastasis: | The unequivocal hallmark of malignancy is the detachment and embolisation of tumour deposits, which effectively spreads disease from one body site to another, usually via the bloodstream or lymphatics. Metastases or secondary tumours are thus discontinuous with the primary tumour and are the major cause of death from malignant disease |
It is now recognised, of course, that cancer is fundamentally a genetic disorder caused by the accumulation of multiple mutations in cellular DNA and, whilst the concepts of initiation, promotion and progression remain relevant, it is really a disease that is characterised by the complexity of its aetiological mechanisms. It is probable that the precise balance of intrinsic genetic abnormality and extrinsic carcinogenic influence is extremely variable and thus perplexingly different in every patient.
Genetics is by strict definition the scientific study of heredity, so it is effectively the field of epigenetics that is of most interest to us in our studies of tumour biology and carcinogenesis. This is because it is particularly the latter science that aims to identify the detailed mechanisms by which individual genes bring about their recognisable or phenotypic effects and tries to determine how such pathways are distorted during cancer formation.
Modern concepts of carcinogenesis, of course, recognise the over-riding importance of molecular genetics. The mutations fundamental to cancer development are those that ultimately produce immortal cells that no longer respond to the normal intracellular and/or extracellular regulatory signals that control cell proliferation, differentiation and death.
The annual estimated world incidence for combined intra-oral and oropharyngeal cancer is around 400 000 cases (making it the sixth most common cancer), with approximately 5000 new cases of oral cancer arising in the UK each year [1].
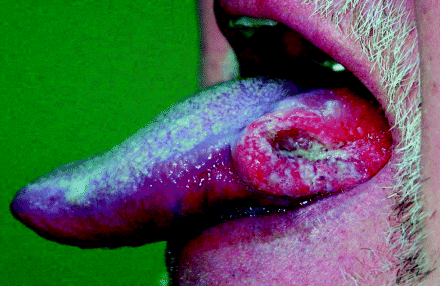
Over 95% of oral cancers are squamous cell carcinomas, originating from keratinocytes of the oral cavity’s stratified squamous epithelial lining. They present most commonly as non-healing ulcerations due to tissue destruction, induration due to excessive keratin formation within tumour stroma, fixation to underlying tissues due to submucosal invasion and raised, everted tumour margins. Figure 3.2 shows the characteristic clinical appearance of a squamous carcinoma arising on the lateral tongue surface.
Figure 3.2 An exophytic squamous cell carcinoma arising from the lateral surface of the tongue showing classic raised, everted margins surrounding a central, cavitating ulceration.
It is probably not surprising that oral epithelial cells are so vulnerable to cancer change, acting as they do as a defensive barrier against irradiation, pollutants in the air we breathe and carcinogens in the food and drink we consume. The fact that many carcinogens are ingested in large quantities, such as those in tobacco products and alcoholic beverages, only compounds the risk for the oral epithelium.
Essentially, a progressive stepwise mutation is believed to lead to a premalignant stage followed by malignant transformation, exhibited by increasing cellular dysregulation, growth abnormalities, cell immortality and ultimately invasive and metastatic malignant behaviour. The concept of an oral premalignant or pre-invasive state is based upon many longitudinal studies in which a number of identifiable oral precursor lesions have been seen to undergo malignant transformation, the observation that many premalignant lesions coexist with established carcinomas, and the fact that both premalignant and malignant disorders share common histopathological and/or biomolecular changes [2].
As we have previously discussed in Chapter 1, it is surprisingly difficult to determine accurate figures for either the overall incidence or prevalence of oral potentially malignant disorders, owing to considerable differences in the patient populations studied, variations in ethnicity and indigenous tobacco habits, and a lack of clarity for both clinical and histopathological definitions of lesions studied. Nonetheless, it is probably accepted that the estimated global prevalence runs somewhere between 2% and 3% [3].
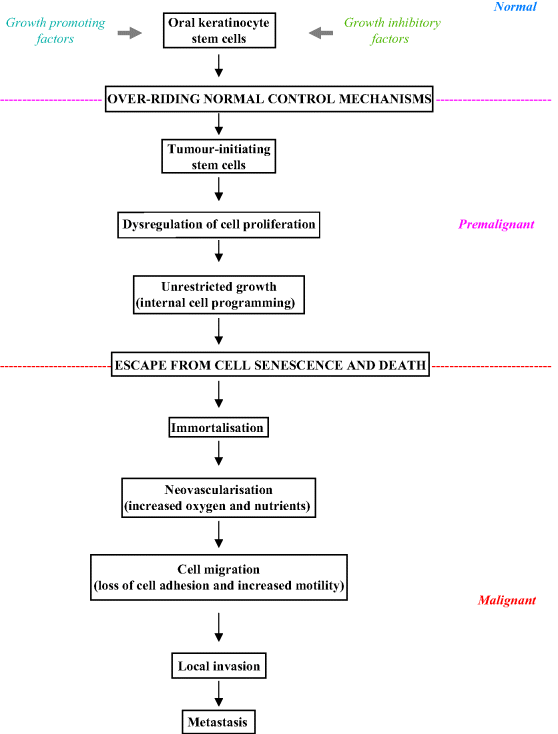
Figure 3.3 summarises the various steps involved during oral carcinogenesis and the potential pathway that a keratinocyte may take through premalignancy to cancer. Cancer growth probably arises from clonal expansion and aberrant growth of a single stem cell or from a few tumour-initiating cells that acquire self-renewal capacity. Stem cells escape normal growth control, as a result of which they gain a significant growth advantage, allowing the development of an expanding clone which then displaces normal epithelial tissue. As the resulting lesion enlarges, additional genetic damage probably gives rise to numerous subclones. Subsequent clonal divergence and selection in such an inherently unstable field eventually gives rise to an invasive cancer [4].
Figure 3.3 Steps in oral carcinogenesis.
As the tumour grows and develops, the genetic instability of the cancer may increase considerably, resulting in a heterogenous tumour cell phenotype, as discussed in the introduction above. Where there is a predominance of resistant, rapidly proliferating cells, this will clearly confer a significant advantage in terms of growth and survival.
Oral cancer cells will also be able to evade apoptosis by inactivation of apoptosis-inducing genes or by enhanced activity of anti-apoptosis genes thereby allowing cell survival despite DNA damage, hypoxia and lack of nutrition. A variety of cell membrane factors, the anti-apoptotic action of BcL-2 proteins in mitochondria, and molecules within the cytosome – such as the inhibitor of apoptosis protein (IAP), survivin and ubiquitin – may all be involved in the evasion of apoptosis during carcinogenesis [5].
There also appears to be a significant increase in angiogenesis and overall vascularity in oral tissue during the transition from normal mucosa through increasingly dysplastic tissue to invasive carcinoma. Angiogenesis is known to be essential for tumour progression and metastasis and it is perhaps not surprising that increased angiogenesis accompanies both increased cell proliferation and decreased apoptosis as histological abnormalities increase in the mucosa [6, 7].
What remains frustratingly difficult, however, is not only our inability to identify individual patients and oral lesions at risk of oral cancer change, but also our lack of any predictive ability to determine the nature and progression of this pathway to malignancy in the clinical situation.
The ‘Progression Model’ for Oral Cancer
Oral squamous cell carcinoma is thus regarded as a genetic disorder that develops from a series of stepwise mutations, sometimes spontaneously but more often than not due to some external carcinogenic influence. This leads to cellular changes that transform normal epithelium into invasive neoplasms. Fundamental to this process are alterations in the genes regulating cell division, cell cycle progression and DNA synthesis and repair. Promotion of oncogenes, inactivation of tumour suppressor genes and alteration of growth factor activity may all lead to abnormal regulation of cell proliferation, which as we have seen in Chapter 2, is recognised as a fundamental hallmark of carcinogenic change.
Oncogenes are derived from normal cellular gene products (proto-oncogenes) that have undergone mutation (point mutations, amplifications or rearrangement of DNA sequencing), conferring increased malignant potential. Tumour suppressor genes are believed to perform protective inhibitory roles at the cellular level, with mutation leading to loss of inhibitory control of cellular proliferation and loss of apoptosis (programmed cell death).
Box 3.2 summarises the many and varied proposed molecular mechanisms and resultant possible biomarkers involved in oral carcinogenesis. These involve multiple genetic and epigenetic events, including abnormalities in cell signalling, growth, survival, cell cycle control and angiogenesis, which together herald the progressive acquisition of a malignant phenotype [8, 9]. It is important to emphasise that many of these processes overlap quite extensively and that they should not, therefore, be regarded as discrete mechanisms. The sequential order in which mutations arise is probably less important than their cumulative number and whilst the accumulation of such abnormalities may sometimes be due to random mischance, they are more commonly caused by environmental carcinogens, especially factors such as tobacco and alcohol.
Box 3.2 Mechanisms and markers of biomolecular disruption in oral carcinogenesis.
Oncogenes
- Signalling pathways:
Epidermal growth factor
Epidermal growth factor receptor
Transforming growth factor alpha
- Transcription factors:
Transcripting factor-activating protein
- Cyclins and cyclin-dependent kinases
Tumour suppressor genes
- p53
- Retinoblastoma protein
Genetic abnormalities
- DNA ploidy:
Aneuploidy
Allelic instability
- Chromosome abnormalities:
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


