CHAPTER 28 Antihypertensive Drugs
About 29% of Americans have hypertension, and it is the leading cause of cardiovascular disease worldwide.10 Careful examination of the prevalence of hypertension reveals that it is distributed disproportionately among subgroups in the U.S. population. Hypertension increases with advancing age, but its prevalence is much lower in women before menopause than in men of comparable age. There also seems to be a racial component to hypertension3: the prevalence is approximately 23% in whites and Mexican Americans but is 32% in African Americans.
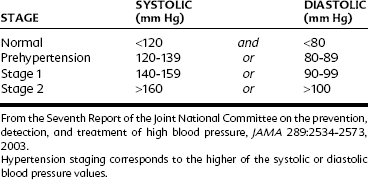
Because of the asymptomatic nature of this disease, approximately one third of affected individuals are unaware of their condition. Isolated systolic hypertension affects more than 15% of all people older than 60 years. Studies suggest that only half of hypertensive patients receive pharmacologic treatment at all, and, of this fraction, only half have adequate control of blood pressure levels. Because the long-term consequences of hypertension (e.g., coronary artery disease, stroke, renal failure) are so devastating to health, screening programs are essential to detect the disease early so that treatment can be instituted before major complications ensue. Education of patients is also essential to ensure compliance with recommended therapy because of the insidious nature of the disease and because unpleasant side effects of the drugs used to treat it may cause the patient to feel better when not receiving medication. An individual is considered hypertensive if his or her systolic or diastolic arterial blood pressure (or both) is elevated above normal (i.e., systolic arterial pressure >140 mm Hg or diastolic arterial pressure >90 mm Hg).2
CLINICAL ASPECTS OF HYPERTENSION
Classification
The severity of hypertension is classified as shown in Table 28-1. Hypertension can arise as a primary disease or as a result of an underlying illness. Essential hypertension is a term used to describe the presence of sustained, elevated blood pressure for which no cause is apparent. When this term was coined, it was believed that the elevation of blood pressure was essential to maintain organ perfusion in the affected patient. This idea is no longer widely accepted, but the term is still in use. Essential hypertension remains of unknown etiology and represents 80% to 90% of all cases of hypertension. Although much is known about the cardiovascular changes that occur as a consequence of prolonged elevation of blood pressure, no single pathologic change can be cited as the primary cause. Many theories abound regarding the causes of essential hypertension, some of which are discussed subsequently.
Secondary hypertension results from a known disorder, such as renal, vascular, or parenchymal disease, or from an endocrine disorder such as pheochromocytoma. Treatment of secondary hypertension usually consists of therapy for the underlying disease process. Hypertension may be systolic or diastolic, or both. Until more recently, less emphasis had been placed on the importance of systolic hypertension. More recent evidence indicates, however, its close association with untoward outcomes.23 Treatment of isolated systolic hypertension in elderly patients with an antihypertensive drug has been shown to reduce mortality rates, especially from stroke.14,19 The results are independent of mean arterial pressure. The risk of heart failure is also decreased with reduction in systolic hypertension.13 It has been suggested that the incidence of dementia is reduced as systolic pressure is reduced in elderly patients.16
In metabolic syndrome, hypertension is accompanied by abdominal obesity, hyperlipidemia with atherosclerosis, and hyperglycemia or insulin resistance or both.22 In this syndrome, hypertension is only one target of therapy. Questions that are still being explored are to what extent each sign of the syndrome is related and how.
Regulation of Blood Pressure
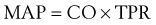
CO is determined by the load presented to the heart (venous return or preload) and the inotropic and chronotropic state of the myocardium. TPR depends on the diameter and compliance (stiffness) of the arterioles. These factors are regulated by the resting vascular smooth muscle tone, intrinsic reactivity of the vasculature, vasoactive substances in the blood, and sympathetic nervous system activity. Another important factor in the governance of blood pressure is the blood volume, which is regulated by the kidneys. The interrelationships among all these factors are illustrated in Figure 28-1.
Long-term stresses on the maintenance of blood pressure (e.g., alterations in water and salt intake) are handled by the kidneys. A change in blood pressure is sensed by the kidneys as a corresponding change in renal perfusion pressure. This disturbance invokes two compensatory mechanisms. First, the tubular reabsorption of Na+ and water either decreases (in high perfusion pressure) or increases (in low perfusion pressure). This alteration adjusts blood volume and secondarily changes CO to bring blood pressure back to normal. The kidneys also influence resistance vessel tone more directly by releasing renin (activating the renin-angiotensin system) when renal perfusion is diminished. The resultant increase in vasoactive angiotensin peptides increases peripheral vascular resistance by causing vasoconstriction. Angiotensin peptides also promote volume retention by increasing the release of aldosterone and contribute to muscular hypertrophy and other structural changes in the heart and vasculature (collectively referred to as remodeling).4
Pathophysiologic Characteristics of Essential Hypertension
The physical findings of a patient with essential hypertension usually reveal that CO is normal and TPR is elevated. In a hypertensive patient, the baroreceptor reflexes function normally, but have been “reset” to maintain MAP at a higher than normal value. The reasons for this shift are not yet understood and are the subject of intensive research. It is evident that there is a genetic component to essential hypertension and that certain risk factors lead to a worsening of blood pressure elevation (Box 28-1). In many patients, long-term cardiovascular complications of hypertension can be controlled solely by making appropriate lifestyle changes.
Although the cause of essential hypertension is unknown, it is well established that high blood pressure leads to cardiovascular and renal disease. Elevated blood pressure is directly correlated with overall mortality (Figure 28-2). It is accepted that reducing the blood pressure in hypertensive patients reduces the risk of cardiovascular events including myocardial infarction and stroke, and kidney failure.23,26 The damage caused by decades of elevated arterial pressure can be seen in the form of left ventricular hypertrophy, medial thickening of arteries, and nephropathy.18,26 These changes contribute to the development of diseases such as congestive heart failure, coronary artery disease, stroke, aneurysm, and renal failure (Box 28-2). Numerous clinical trials have shown a reduction in morbidity and mortality rates after pharmacologic reduction in blood pressure in hypertensive patients.
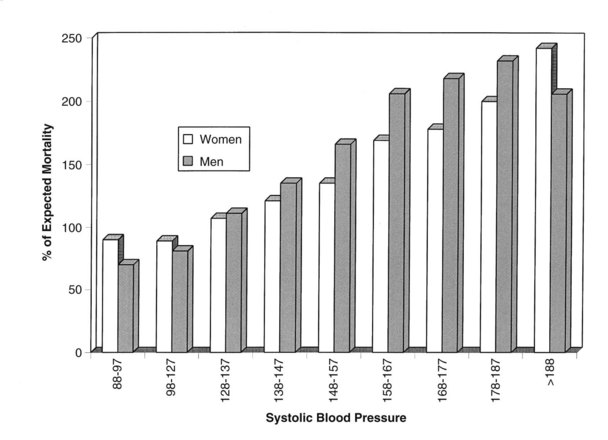
FIGURE 28-2 Expected mortality as a function of systolic blood pressure at all ages without regard to treatment.
(Adapted from The Society of Actuaries and the Association of Life Insurance Medical Directors of America: Blood pressure study 1979, Boston, 1980, The Society.)
Diabetic patients are particularly vulnerable to targeted organ damage resulting from hypertension. The current standard of care dictates that antihypertensive therapy be prescribed in diabetics whose blood pressure is in the high-normal range or above (see Table 28-1). Angiotensin-converting enzyme (ACE) inhibitors are most commonly used for this purpose because of their well-documented protective effects in diabetic patients.4,21
General Aims of Antihypertensive Drug Therapy
Treatment of essential hypertension consists of therapy aimed at reducing the blood pressure into the normal range. As shown in Figure 28-1, many factors play a role in the determination of blood pressure, and consequently pharmacologic agents with diverse mechanisms of action can be used singly or in combination to treat essential hypertension. Antihypertensive agents can be categorized according to their mechanism of action and therapeutic use: diuretics, drugs affecting angiotensin, Ca++ channel blockers (CCBs), drugs affecting sympathetic function, direct-acting vasodilators, and miscellaneous drugs. Because the basic pharmacologic properties of many drugs useful in treating hypertension are discussed elsewhere, only pharmacologic features pertinent to the treatment of hypertension are discussed in detail in this chapter. Figure 28-3 shows the major sites of action of antihypertensive agents.
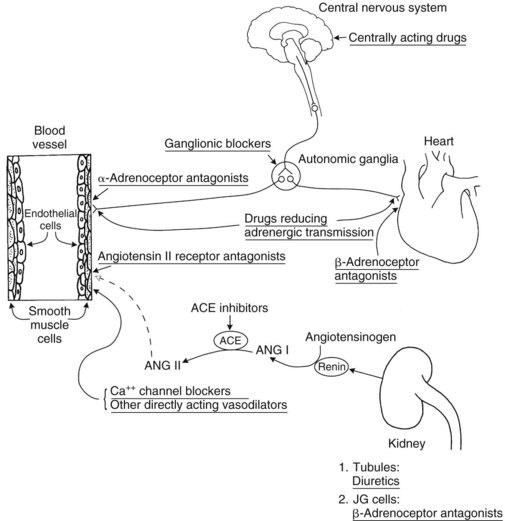
DIURETICS
Diuretics reduce plasma volume by increasing Na+ and water excretion. Initially, this effect reduces blood pressure by decreasing CO. With time, CO and extracellular fluid volume return toward normal values, but the hypotensive effect persists because of a reduction in peripheral resistance. It is probable that electrolyte changes in vascular smooth muscle account for the vasodilation. For a complete discussion of diuretics used in the treatment of hypertension, see Chapter 27.
DRUGS AFFECTING ANGIOTENSIN
The role played by the renin-angiotensin system in hypertension has received much attention in recent years.4,25 Renin catalyzes the conversion of angiotensinogen, a glycoprotein found in the blood, to angiotensin I, a decapeptide with little cardiovascular activity (see Figure 28-3). Angiotensin I is activated by conversion to the octapeptide angiotensin II. This reaction is catalyzed by ACE, otherwise known as dipeptidyl carboxypeptidase or peptidyl dipeptidase. Under its designation as kininase II, ACE is also the enzyme that inactivates bradykinin.
Angiotensin-Converting Enzyme Inhibitors
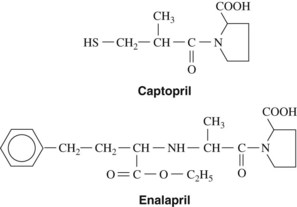
ACE inhibitors are among the most commonly used drugs for the treatment of essential hypertension. Captopril, the first drug of this class to be developed, was specifically designed to disrupt the renin-angiotensin pathway. Its structure is shown in Figure 28-4. Captopril differs from other ACE inhibitors because it contains a sulfhydryl group. Enalapril, lisinopril, and most of the other ACE inhibitors have an amino acid substitution. Fosinopril contains a phosphorus linkage.
Pharmacologic effects
Drugs that inhibit ACE block the conversion of angiotensin I to angiotensin II (Figure 28-5; see Figure 28-3).4 ACE inhibitors markedly decrease blood concentrations of angiotensin II and induce an immediate decrease in blood pressure. They may also act to maintain the lowered blood pressure by elevating bradykinin (a potent vasodilator) concentrations in the blood (see Figure 28-5). (As previously mentioned, ACE, as kininase II, is responsible for the breakdown of bradykinin.) ACE inhibitors have an antihypertensive effect even in patients without high renin activities. Over the course of several weeks, blood pressure is progressively reduced, mainly through decreased peripheral resistance, with little effect on CO or renal blood flow. Salt and water retention is not induced, and orthostatic hypotension and tachycardia are not problems.
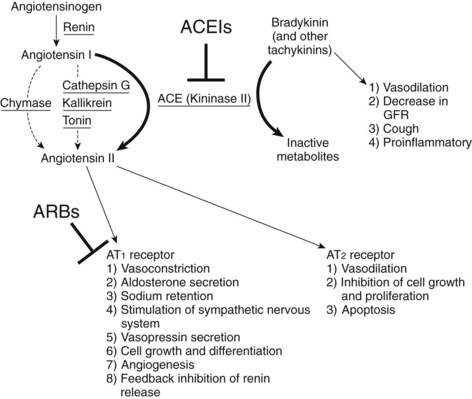
The reduction in angiotensin II concentrations as a result of ACE inhibition leads to a decrease in aldosterone secretion, which results in an increase in Na+ and water excretion. Correspondingly, there is a net increase in the reabsorption of K+ in the kidney tubule. Hypokalemia is not an adverse effect of ACE inhibitors. K+ supplements and K+-sparing diuretics should not be used concurrently with ACE inhibitors to avoid hyperkalemia. With long-term ACE inhibitor therapy, deleterious cardiovascular remodeling may be reduced or even reversed.4,5,26 ACE inhibitors are also renoprotective and because of this are useful drugs in patients with chronic renal disease and diabetes. The presence of high normal (or above normal) blood pressure and diabetes is a clear indication for the use of an ACE inhibitor.
Absorption, fate, and excretion
Enalapril (see Figure 28-4) is a prodrug that must be hydrolyzed in the liver to become fully active. Its absorption is not influenced by food, and it has a longer duration of effect than captopril. The active metabolite enalaprilat, with a plasma half-life of 11 hours, provides for the extended action. Enalaprilat is not absorbed from the gastrointestinal tract but is effective after intravenous administration; it has been marketed for such use in patients unable to take drugs orally. Other ACE inhibitors with an esterified carboxyl side chain are also activated (by hydrolysis) in the liver to “prilat” metabolites that avidly bind to ACE and provide durations of effect sufficient for single daily dosing. The other ACE inhibitors are listed at the end of this chapter. They differ primarily in their pharmacokinetic properties.
Adverse effects
The most frequent side effect of the ACE inhibitors is coughing, which occurs in 20% of patients.4 Altered or reduced taste sensation is also common, especially with captopril. These adverse effects may disappear after continued use. The significance of reported cases of proteinuria is not established at this time. Other adverse effects that have been documented are skin rash; angioedema of the face, mucous membranes of the mouth, or extremities; and flushing, pallor, and hypotension. Angioedema is a serious condition that demands withdrawal of the drug. Hyperkalemia and neutropenia may rarely occur.
ACE inhibitors may cause renal insufficiency in patients with bilateral renal stenosis. The mechanism is the reduction of renal angiotensin II production, leading to a disproportionate dilation of efferent renal blood vessels compared with afferent vessels. This vascular imbalance results in a significant decline in the glomerular filtration rate. ACE inhibitors can help preserve renal function in diabetic patients.4,26 ACE inhibitors have the beneficial effect of reducing proteinuria in patients with some renal diseases.
Angiotensin Receptor Antagonists
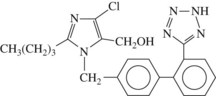
Losartan (as losartan potassium) was the first orally active angiotensin II receptor antagonist to be introduced (Figure 28-6). Other angiotensin II antagonists include candesartan, eprosartan, irbesartan, telmisartan, and valsartan. These nonpeptide analogues of angiotensin bind to the angiotensin II receptor and competitively inhibit the action of angiotensin II and angiotensin III.4,6 They are selective inhibitors of the AT1 receptor, the angiotensin receptor subtype that accounts for the major physiologic effects of angiotensin II. The effect is to inhibit the consequences of AT1 receptor stimulation without affecting potentially beneficial effects mediated by the AT2 receptor (see Figure 28-5).23 As is the case with ACE inhibitors, AT1 receptor blockers reduce the blood pressure and the tissue remodeling seen in hypertension and reduce organ damage resulting from hypertension.4,15,26
Losartan has a half-life of only 1.5 hours, but it is metabolized to an active metabolite with a longer half-life. Valsartan has a plasma half-life of 6 hours; it is excreted in the bile largely in the unchanged form. The half-lives for the other angiotensin II blockers range from 6 hours for eprosartan to 24 hours for telmisartan. The selectivity of angiotensin II antagonists avoids some of the side effects of ACE inhibitors, such as coughing and angioedema, because the bradykinin pathway is not affected by the angiotensin II antagonists (see Figure 28-5). Orally effective angiotensin II receptor antagonists now constitute a major drug group for treating hypertension.5
Renin Inhibitors
The renin inhibitor aliskiren has been approved for use in the United States as a once-daily treatment for stage 1 hypertension (see Table 28-1). Aliskiren binds with high specificity to the proteolytically active site of human renin.21 Renin is the rate-limiting step in the renin-angiotensin system, so inhibition of this enzyme provides a logical control point for pharmacologic intervention. Aliskiren reduces circulating concentrations of angiotensin I and angiotensin II, producing a decrease in systolic and diastolic blood pressure, comparable to reductions seen with ACE inhibition or AT receptor antagonism. Early reports on toxicity are favorable. Based on the mechanism of action, expected adverse events include hyperkalemia and hypotension. Aliskiren administration produces hyperreninemia owing to a compensatory increase in renin release. This effect is not clinically significant. Aliskiren does not interfere with ACE-induced catabolism of bradykinin, and it is not expected that cough or angioedema would be produced by this class of drugs (as seen in ACE inhibitors). Similar to other inhibitors of the renin-angiotensin system, aliskiren is contraindicated in patients with bilateral renal artery stenosis and during pregnancy. Aliskiren has a poor bioavailability and is greater than 90% excreted unchanged in the feces, so minimal drug metabolism interactions are expected from this drug.
CA++ CHANNEL BLOCKERS
Verapamil, diltiazem, and nifedipine were the first CCBs to be marketed. The pharmacologic features of nifedipine and its dihydropyridine congeners, including amlodipine, felodipine, isradipine, nicardipine, nimodipine, nisoldipine, and nitrendipine, are addressed in this chapter. Other CCBs are discussed in Chapters 24 and 26.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


