CHAPTER 23 Drugs for Treating Orofacial Pain Syndromes
PHARMACOLOGY AND CHRONIC PAIN
The management of chronic orofacial pain, as compared with acute pain, requires an in-depth knowledge of pharmacology and pharmacotherapy because chronic pain disorders are a heterogeneous group of conditions with various pathologic mechanisms and characteristics requiring diverse families of medications for treatment. Dentists do not generally use these medications because dentistry has traditionally focused on acute pain problems. The pharmacologic characteristics of opioids are discussed in Chapter 20, and the pharmacologic characteristics of acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs) are discussed in Chapter 21. Treatment of acute pain in dentistry is addressed in Chapter 47.
SEROTONIN (5-HYDROXYTRYPTAMINE)
Historical Aspects of Serotonin
Janeway and associates20 did a thorough investigation of the vasoconstrictive substance and noted that it was not present in uncoagulated or citrated blood, that it was definitely associated with platelets, that it was soluble in water more than ether or chloroform, and that the factor did not depend on the clot formation but on the disintegration of the platelets in the clot. The substance itself was eventually isolated and named serotonin by Rapport and colleagues in 1948.36 Shortly after this, Rapport and colleagues identified the agent as 5-HT, and Hamlin and Fischer18 reported synthesizing it in 1951.
Meanwhile, in Italy, in a separate series of studies, Erspamer and Asero8 isolated a substance from the mucosa of rabbit stomach and found that it was abundant in the enterochromaffin cells of the gut, could be extracted with alcohol and acetone, was an amine that affected smooth muscle, and was deactivated by deamination. Erspamer and Asero named it enteramine. By 1952, serotonin and enteramine had been chemically identified as 5-HT, eventually leading to international wrangling over the naming of 5-HT. It was argued that “enteramine” was inaccurate because the substance was found in places other than the gut, and “serotonin” was equally inadequate from the points of origin and pharmacologic action. In 1986, when the International Serotonin Club was organized, American researchers prevailed over the European contingent in naming the substance serotonin by arguing that serotonin was the most widely accepted name, 5-hydroxytryptamine was too long, and 5-HT was only an abbreviation (but one used here).
5-HT and Pain
Stimulation of the periaqueductal gray (PAG) was shown to modulate nociception on a spinal level.28 This effect is known as stimulation-produced analgesia (SPA). Although a number of areas have been studied in animals, human studies of necessity have been limited. In humans, stimulation of the midbrain region of the PAG and areas slightly more rostral in the periventricular gray matter of the hypothalamus are known to produce SPA. Neurosurgeons were able to show SPA in humans by stimulating the equivalent human midbrain sites. Researchers had determined that electrical stimulation of brainstem PAG produced analgesia in animals. Although the exact boundaries of the responsive area were not clearly defined, the sites most responsive to SPA were: ventral to the midbrain cerebral aqueduct; in the PAG; sites lateral to this structure; the rostroventral medulla (RVM), including the midline nucleus raphe magnus (NRM) and reticular formations; the hypothalamus; the frontal lobe; and the spinal cord. Areas outside of the midbrain have not been systematically studied.
Anatomic Distribution
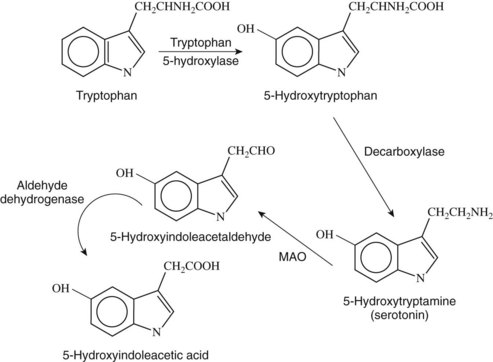
5-HT is a biogenic monoamine and is widely distributed throughout the plant and animal kingdom. In mammals, the highest concentrations are found in the enterochromaffin cells of the gastrointestinal mucosa, central nervous system (CNS), and blood platelets. The structure of 5-HT is shown in Figure 23-1. Its most notable features are the hydroxyl group on position 5 of the indole nucleus and the primary amine nitrogen that can accept a proton, making the compound hydrophilic and unable to pass the blood-brain barrier easily. Rapport and colleagues36 found the substance in the brain, indicating that it must be synthesized and perform some unidentified function there. It was subsequently assumed that 5-HT was associated with psychiatric disorders such as depression and schizophrenia when it was shown that the psychedelic drug lysergic acid diethylamide (LSD) antagonized 5-HT function. 5-HT is now known to be involved in many behavioral and psychiatric disorders, such as schizophrenia, obsessive-compulsive disorder, depression, and anxiety, and drugs that have an effect on the 5-HT system have been beneficial in treating these disorders (see Chapter 12).
Despite earlier suggestions that 5-HT was a neurotransmitter synthesized in the brain, the actual localization of 5-HT neurons was not determined for at least 10 more years. By using lesioning and fractionation techniques, 5-HT was grossly associated with specific neuronal elements, but it was impossible to observe the relationship directly until fluorescence histochemical techniques were developed. This process had inherent problems, however, that made identification a significant challenge. Dahlström and Fuxe,7 using immunocytochemical techniques, localized 5-HT–associated neurons in nine discrete clusters of cells along the midline of the upper brainstem and pons. These 5-HT–containing cell bodies, designated B1 to B9, corresponded for the most part to the dorsal raphe nuclei. Only approximately 40% to 50% of the dorsal raphe nuclei are serotonergic neurons, and some serotonergic nuclei are found outside the midline raphe nuclei area, although the major brain concentration is in the dorsal raphe nuclei.
Additional studies have shown that the lateral and dorsolateral pontine tegmentum, which contain many noradrenergic neurons, are also involved in nociceptive modulation when stimulated, and these sites send projections to the PAG, the RVM, and the spinal cord. The projections from the lateral and dorsolateral pons are noradrenergic and possess important α2-adrenergic receptors. In animal studies, norepinephrine (NE) applied directly to the spinal cord blocks response to nociception through selective inhibition of the nociceptive dorsal horn neurons (see Chapter 20). Lesioning the white matter of the dorsolateral funiculus of the spinal cord blocks the inhibitory effect of SPA and confirms the existence of a descending modulatory pathway that travels through the dorsolateral funiculus. Further studies of the dorsolateral funiculus projections to the spinal cord have found that most of the brainstem projections arise in the RVM and dorsolateral pons, with few projections from the PAG. This finding implies that the PAG projections must be relayed through the RVM. This has been confirmed by studies showing that the major neuronal input to the RVM is from the PAG and adjacent structures, and lesioning or blocking RVM cells eliminates the analgesic effect obtained from PAG stimulation.
Anti–5-HT antibody labeling has identified 5-HT in all dorsal horn laminae, but the highest densities are found in laminae I, II, IV, V, and X. The RVM projections terminate mainly in laminae I, II, and V. These areas are important for pain because this is where the central terminals of afferent nociceptors and cell bodies of second-order neurons are found. This dorsal horn area is the major “switchboard” for pain, and stimulation of the PAG and RVM modulates nociceptive activity here (see Chapter 20).
Transmission of sensory and particularly nociceptive messages by afferent fibers entering the dorsal horn of the spinal cord is under control of pathways originating in the ventromedial medulla. It had been observed that neurons from the medullary raphe nuclei and particularly the NRM project predominantly to the dorsal horn, including the superficial laminae and the area around the central canal, and are involved in a descending inhibitory pathway for modulation of nociceptive input. Because the area was found to have an abundance of 5-HT–containing neurons, researchers postulated that 5-HT was a descending pain modulatory system neurotransmitter. 5-HT–containing neurons are located in the rostroventromedial medulla and caudal pons, and particularly in the NRM, the nucleus paragigantocellularis, and the ventral portion of the nucleus gigantocellularis. More recent studies have described other descending projections from the bulbomesencephalon to the spinal cord that do not contain 5-HT and are more numerous within the medulla and caudal pons, indicating that descending modulation is not limited to 5-HT fibers.24
Immunocytochemical studies of antibodies directed against 5-HT have shown that two distinct types of 5-HT neurons innervate the cerebral cortex of many mammals. The studies have found fine axons with small varicosities originating from the dorsal raphe nuclei and beaded axons with large spherical varicosities originating from the median raphe nuclei. Apparently the two types of axons have different regional and laminar distributions and exhibit different sensitivities to neurotoxic drugs such as 3,4-methylenedioxymethamphetamine, commonly referred to as “ecstasy.” The fine axons seem to be more sensitive to the neurotoxic effects, with loss of functions that may be long-term or permanent. Cooper and associates6 suggested that laboratory animal findings may relate to humans’ use of the drug because the doses commonly used by recreational drug users are similar to what are used in animal studies. Ecstasy users have shown a 26% decrease in 5-HIAA, the 5-HT metabolite. The decrease in metabolite may indicate a decrease of 5-HT function in the brain related to loss of some 5-HT neurons. The functional distinction between these two types of neurons generally remains unclear, however.
Synthesis, Storage, and Fate
5-HT is synthesized from the amino acid l-tryptophan (see Figure 23-1). Although platelets contain large amounts of 5-HT, it only accumulates rather than being synthesized there. Synthesis in the CNS involves active transport of tryptophan through the blood-brain barrier. Tryptophan is derived primarily from the diet, and its elimination from the diet can profoundly decrease brain 5-HT. In addition, the active transport of tryptophan is affected by its concentration in the blood and the relative concentration of other amino acids that are transported by the same active transport mechanism. l-Tryptophan is converted in serotonergic neurons containing the enzyme tryptophan hydroxylase (l-tryptophan-5-monooxygenase).
The initial synthesis step is hydroxylation of tryptophan at the 5 position to form 5-hydroxytryptophan (see Figure 23-1). Tryptophan hydroxylase, the enzyme responsible for this reaction, occurs in low concentrations in most tissues, including the brain, and has proved to be difficult to isolate.
5-Hydroxytryptophan is rapidly decarboxylated to form 5-HT by the aromatic enzyme l-amino acid decarboxylase, which is the same enzyme that catalyzes the decarboxylation of l-dopamine in catecholamine neurons (see Figure 23-1). Because the rate of the reaction is so rapid and requires less substrate than the initial reaction, the action of tryptophan hydroxylase in the first step is regarded as the rate-limiting step in the synthesis of 5-HT, and drugs targeting the action of the decarboxylase have not been shown to be effective.
Metabolism
5-HT is also metabolized in the liver by the enzyme monoamine oxidase (MAO) (see Figure 23-1). The product of this reaction is 5-hydroxyindoleacetaldehyde, which is oxidized further by aldehyde dehydrogenase to form the final acid metabolite, 5-HIAA, which is excreted (see Figure 23-1). It had been suggested that increased levels of either of the metabolites of 5-HT or the concentration of 5-HT itself would affect its metabolism, but it has been noted that using MAO inhibitors to block metabolism does not affect the synthesis of 5-HT, and concentrations increase to three times greater than controls. If the elimination of 5-HIAA is blocked by the drug probenecid, the 5-HIAA levels continue to increase without apparent feedback inhibition. The implication of these findings is that the synthesis of 5-HT is not affected by changes in concentrations of its metabolites.
Reuptake and transport across body membranes
Presynaptic reuptake of 5-HT from the synaptic cleft is a major mechanism for controlling the synaptic concentration and action of 5-HT. The presynaptic terminals of serotonergic neurons contain high-affinity uptake sites that are involved in this process by using a plasma membrane transporter protein that can transport 5-HT in either direction depending on the concentration gradient. The transporter proteins involved in this process are composed of 12 membrane-spanning proteins that are Cl− and Na+ dependent. Although older TCAs inhibit the reuptake of 5-HT, they also have a variable capacity to inhibit NE reuptake and affect other systems and receptors (see Chapter 12). SSRIs that have shown great utility in moderating depression, anxiety, and obsessive-compulsive disorders also inhibit the 5-HT transporter proteins. These medications are more 5-HT selective with more limited effects on the NE transporter.
5-HT Receptors and Pain
In 1957, Gaddum and Picarelli11 reported two separate 5-HT receptors in peripheral smooth muscle preparations studied in vitro. Since then, there has been an exponential development of information relating to 5-HT receptor types and functions, and numerous receptor subtypes have been identified and cloned more recently. Nevertheless, the complete picture of how 5-HT and its receptors modulate pain remains obscure. There are now seven main family groups of receptors, but current understanding attributes most of the 5-HT actions relative to pain to the families designated 5-HT1, 5-HT2, 5-HT3, 5-HT4, and 5-HT7. Each receptor family is operationally and structurally distinct, each having its own separate transducing system.
5-HT1 receptors
The 5-HT1 family of receptors (Table 23-1) produces its cellular action by inhibiting adenylyl cyclase and opening K+ channels. Binding studies with autoradiographic ligands have shown binding sites throughout the spinal cord gray matter, raphe nuclei, and substantia gelatinosa, with the higher concentrations in laminae I and II of the dorsal horn and lower in lamina VII in the ventral horn. The hippocampus, the substantia nigra, and dorsal raphe contain the highest concentrations.

5-HT1A receptor
Taiwo and Levine42 showed that 5-HT1A receptors are implicated in peripheral mechanical hyperalgesia. They reported that the hyperalgesia could be blocked by selective 5-HT1A antagonists injected locally. It was also shown in various pain models that the 5-HT1-3 receptors all mediated pain. Powell and Dykstra35 suggested that 5-HT1A receptor agonists may reduce the effects of morphine in an electrical shock model for pain. This same effect was not seen with agonists at 5-HT2, 5-HT3, or α2-adrenergic receptors.
5-HT2 receptors
Although the exact role of 5-HT neuronal modulation is still unclear in migraine, the medications that seem to give the most benefit have definite 5-HT activity. The role of 5-HT in platelets during the ictal phase of migraine is apparently important but remains to be defined. All aspects of the 5-HT system seem to come into play during a migraine attack and not only the 5-HT1 receptors, but also 5-HT2 receptors. It has been observed that some of the most commonly used compounds in migraine prophylaxis—propranolol, pizotifen, methysergide, cyproheptadine, amitriptyline, and chlorpromazine—have an antagonistic effect on specific subtypes of the 5-HT2 receptor family, now known as the 5-HT2B/5-HT2C (formerly 5-HT1C) receptors. This hypothesis is supported by previous observations that the 5-HT2 receptor agonist m-chlorophenylpiperazine (m-CPP) triggers migraine in susceptible individuals when it is administered at doses high enough to activate 5-HT2B/2C receptors.21
5-HT4 receptors
In contrast to the inhibitory effects of the 5-HT1 receptors, the 5-HT4 family of receptors is positively coupled to adenylyl cyclase, provoking second messenger activation of cyclic adenosine 3′,5′-monophosphate (cAMP). This receptor has high concentration in the gut, and antagonists of the receptor prevent development of increased bowel activity that would be stimulated by 5-HT or 5-hydroxy-l-tryptophan sensitization. Ghelardini and colleagues13 reported that 5-HT4 agonists had an antinociceptive effect, raising pain thresholds in mice and rats.
Physiologic Function and Drug Intervention
5-HT released from neurons has presynaptic and postsynaptic receptor effects. There are many options for affecting the availability of 5-HT directly, either by inhibiting the processes that decrease its availability or by enhancing the processes that make it available (Figure 23-2). Figure 23-2 shows sites of interaction with known drugs that influence the 5-HT system. Of all the options, reuptake blockade of 5-HT by TCAs is the most common mechanism of 5-HT active medications prescribed for the treatment of depression and chronic pain; however, the issue of availability of 5-HT to help modulate pain has yet to be settled.
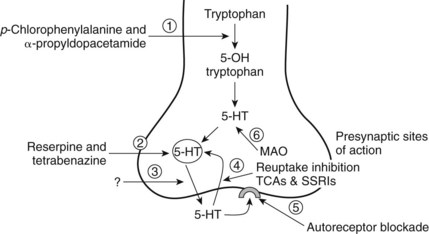
FIGURE 23-2 Possible drug sites for influencing 5-HT neurotransmission. The sites of potential drug action on the 5-hydroxytryptamine (5-HT) neuron are enumerated from 1 to 6. Site 1 represents the modulation of enzymatic action forming the 5-HT precursor and ultimately 5-HT from tryptophan. Site 2 is a potential target for drugs that affect the storage vesicles of 5-HT. Reserpine and tetrabenazine are known to interrupt the storage and cause release of 5-HT. Site 3 targets the release mechanism itself; however, there are currently no known agents that act to interrupt or increase the action of the transporter proteins that carry the 5-HT molecule across the membrane. Site 4 involves the reuptake mechanism that brings 5-HT back into the intracellular environment to be repackaged in the vesicles. Numerous drugs inhibit the reuptake of 5-HT (see Chapter 12). Site 5 refers to the 5-HT autoreceptor. Blockade of this receptor site allows more presynaptic release of 5-HT. Site 6 focuses on the action of monoamine oxidase that converts the free 5-HT to the metabolic product 5-HIAA. Monoamine oxidase (MAO) inhibitors such as phenelzine block this action. SSRIs, Selective serotonin reuptake inhibitors; TCAs, tricyclic antidepressants.
DRUGS FOR ACUTE TREATMENT OF MIGRAINE
Sicuteri and associates40 were the first to note a relationship between 5-HT and migraine in their report on the significant increase in 5-HIAA in the urine of migraine subjects during attacks. Subsequent data did not show a consistent increase in 5-HIAA in all patients with migraine. Nevertheless, the relationship between migraine and 5-HT became solidified at that time and has been elucidated further to the present. Further studies have noted increases in plasma 5-HT, decreases in 5-HT platelet content, and increases in 5-HIAA content in cerebrospinal fluid in migraine patients. These observations support the theory that migraine is caused by chronic 5-HT dysregulation. Further support for the role of 5-HT in migraine has come from clinical positron emission tomography scan studies in patients during migraine attacks showing increased blood flow in the highly serotonergic dorsal raphe nucleus area.
Ergot Derivatives
In the Middle Ages, epidemics of a gangrenous disorder known as “holy fire” or “St. Anthony’s fire” were afflicting communities in Europe. The condition was so named because of the attendant burning experienced by the sufferer. The disorder soon became associated with the grain of rye that had been contaminated with the ergot fungus Claviceps purpurea. In 1918, the ergot alkaloid ergotamine was isolated from the fungus and was found to have sympatholytic activity (see Chapter 7). Shortly thereafter, it was proposed for use as a therapeutic agent for migraine.
Ergot derivatives are nonselective partial agonists and antagonists at 5-HT receptors, having high affinity for the 5-HT1B, 5-HT1D, 5-HT1F, and 5-HT2 receptors and low to moderate affinity for the 5-HT1C and 5-HT3 receptors. It is currently believed that their primary mode of action in alleviating migraine attacks is through their action on the 5-HT1B/1D receptors, inhibiting neurogenic inflammation and nociceptor activity. Table 23-1 lists some 5-HT receptors and related medications and some indications for the drugs.
Ergotamine
Ergotamine is used as an abortive drug at the onset of the migraine attack. Typically, abortive medications have to be taken early in the onset of the migraine because absorption and distribution are impaired as the gastric symptoms of migraine increase. The combination of caffeine with ergotamine speeds gastric absorption, getting the medication into the system more rapidly. In addition to the adverse effects listed in Box 23-1, ergotamine can cause gangrene and damage to blood vessels. The drug is given for short periods at carefully controlled doses.
Dihydroergotamine
Dihydroergotamine comes in a parenteral form and as a nasal spray. The parenteral form can be used intravenously, intramuscularly, or subcutaneously. The nasal form has a bioavailability of less than 65%, which may significantly limit its ability to abort a headache in many patients. This medication has definite advantages over the triptans with short half-life and is associated with lower vasoconstrictive potential. Dihydroergotamine is useful in the hospital to treat protracted unresponsive migraine. Side effects and contraindications for dihydroergotamine, ergotamine, and the other ergot alkaloids are listed in Box 23-1. Methylergonovine and methysergide, semisynthetic ergot alkaloids, are discussed subsequently.
Triptans
Moskowitz29,30 developed the concept of neurovascular inflammation in the trigeminovascular system as a migraine mechanism. He observed that sumatriptan’s action on peripheral neuronal 5-HT1D receptors blocked subsequent release of neuropeptides such as SP and CGRP that were responsible for the development of neurovascular inflammation with concomitant swelling of the dural blood vessels. Moskowitz then proposed that the blood vessel dilation and plasma extravasation noted during the migraine attack was an epiphenomenon of the migraine and not the cause of the migraine, as had been proposed by Graham and Wolff.17
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


