CHAPTER 20 Opioid Analgesics and Antagonists
OPIOID ANALGESICS
The first documented descriptions of Papaver somniferum appeared in approximately 1550 bc in the Ebers papyrus of ancient Egypt.35 Later, in the third century bc, writings of the Greek philosopher Theophrastus contained references to poppy juice. The active analgesic and constipative principle of the poppy plant was not isolated, and the drug was not named (after Morpheus, the Greek god of sleep) until 1806, when Sertürner first isolated morphine from opium and described its properties. Currently available opioid analgesics in addition to morphine and codeine are either semisynthetic congeners of morphine (e.g., hydromorphone, oxymorphone, hydrocodone, and oxycodone) or entirely synthetic (e.g., meperidine, fentanyl, methadone, and propoxyphene).
Basis of Opioid Action
The mechanisms by which opioids act at specific central nervous system (CNS) and peripheral sites to produce their effects are fairly well understood. In the early 1970s, binding sites in the CNS were discovered that stereospecifically, saturably, and reversibly combined with opioids.37 These receptors were later shown to be the natural effectors of opioid action; that is, specific pharmacologic effects were produced by binding of opioids to these receptors. The discovery of opioid receptors naturally raised questions about their biologic significance, spurring research that led to the discovery of endogenous opioid receptors and peptides. Subsequently, several families of endogenous opioid peptides and numerous opioid receptors have been characterized.
Endogenous opioid peptides
There are four families of endogenous opioid peptides: endomorphins, endorphins, enkephalins, and dynorphins. Figure 20-1 illustrates their biologic derivations and structural relationships. The pentapeptide enkephalins—methionine-enkephalin (met-enkephalin) and leucine-enkephalin (leu-enkephalin)—were the first endogenous opioids to be discovered.13 These peptides were subsequently shown to be potent opioid receptor agonists in the same biologic systems in which morphine is active. Initially, the enkephalins were thought to be derived from a larger 91-amino acid peptide, β-lipotropin, but it is now clear that β-lipotropin gives rise to a separate group of opioid peptides, the endorphins (see later).
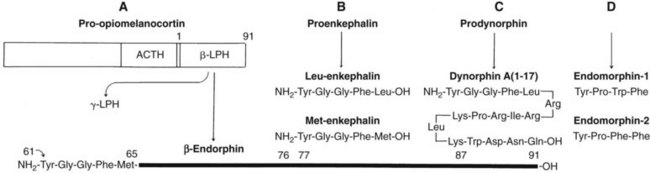
Endomorphin-1 and endomorphin-2 are newly discovered endogenous opioid peptides.47 Both are short tetrapeptides (NH2-Tyr-Pro-Trp/Phe-Phe-CONH2) and structurally distinct from the other opioid peptides (see later). The endomorphins have been localized to areas in the CNS associated with pain processing (e.g., spinal dorsal horn, trigeminal nucleus, midbrain periaqueductal gray) and in the endings of sensory neurons that terminate in the spinal dorsal horn. Although the genes for the endorphins, enkephalins, and dynorphins are known, the gene for the endomorphins has yet to be isolated.
Sites in the CNS where opioid peptides are located differ for the different peptides, confirming that they are involved in different functions. It is a mistaken impression that all opioid peptides in all locations are involved in the modulation of pain. Neurons containing endomorphins are not as widely distributed in the CNS as neurons containing other opioid peptides, but endomorphins are located in pain-processing areas where they are considered to function as neurotransmitters that act at opioid receptors. Neurons containing enkephalins are widely distributed throughout the brain (e.g., striatum, limbic system, midbrain, and medulla) and the spinal cord where they, too, are considered to function principally as neurotransmitters.22 Prodynorphin-derived peptides are abundant in the pituitary, hypothalamus, midbrain, and striatum. Differential processing of proenkephalin and prodynorphin in various brain areas leads to different products having different functions.
Opioid receptors
Three opioid receptors have been cloned: mu (μ), kappa (κ), and delta (δ) (Table 20-1).15,32 They share considerable structural homology (approximately 60% to 65%), contain seven membrane-spanning α-helical segments, and are coupled to transducing G proteins (Figure 20-2). G proteins couple opioid receptors to intracellular effectors and exist as heterotrimers; there is structural and functional diversity in the three G protein subunits. Each heterotrimer consists of an α subunit isoform (of which there are at least 18) and a dimer of β-γ subunits (which also exist in multiple isoforms) that link with specific (and potentially diverse, given the numbers of isoform combinations that are possible) effector systems. Specifically, opioid receptors couple in this way with adenylyl cyclase and ion channels to reduce neurotransmitter release (see later).
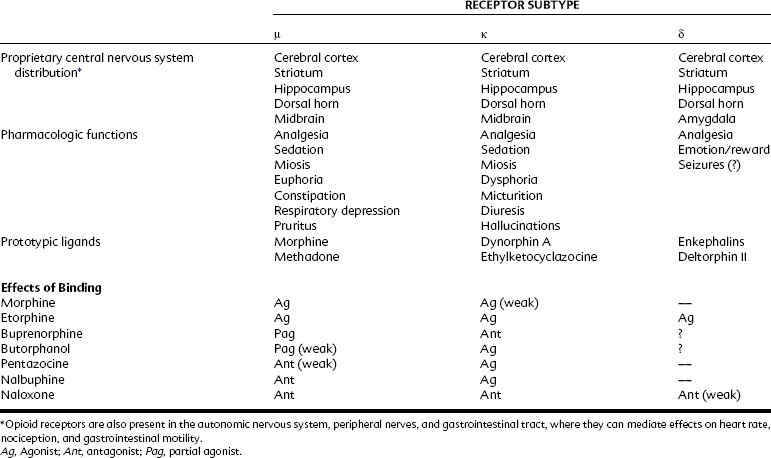
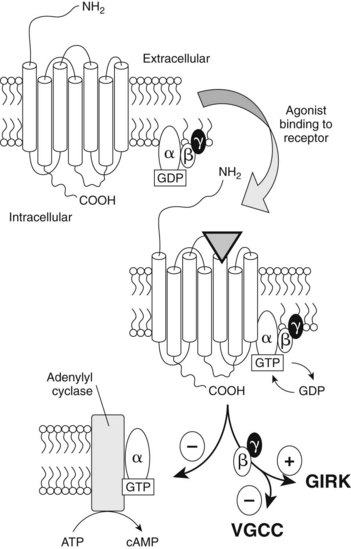
FIGURE 20-2 Diagram of a G protein–coupled opioid receptor. Opioids bind within the hydrophobic membrane-spanning domains of the receptor. Opioid agonist effects at the receptor are mediated by G proteins, an α subunit associated with guanosine diphosphate (GDP), and a β-γ dimer. When an opioid agonist binds, the conformation of the receptor is changed as the membrane-associated G proteins assemble with the receptor. GDP is exchanged with guanosine triphosphate, and this activated Gα complex negatively regulates adenylyl cyclase. The β-γ dimer activates conductance in a G protein inwardly rectifying K+ (GIRK) channel and inhibits voltage-gated Ca++ channels (VGCC) in the cell membrane. (See Chapters 1 and 5 for more details on signal transduction.) ATP, Adenosine triphosphate; cAMP, cyclic 3′5′-adenosine monophosphate.
The amino acid sequences of the three opioid receptors are most homologous in the transmembrane domains and intracellular loops; the sequences share little homology in the extracellular loops and N- and C-termini. Each receptor is distributed differently in the CNS, peripheral nervous system, and smooth muscle of the gastrointestinal tract. A receptor with high sequence homology to the three cloned opioid receptors, termed ORL-1 (for opioid receptor–like, an “orphan” receptor), has been discovered.21,33 Despite its structural similarity to opioid receptors, opioids do not bind to ORL-1 with high affinity. A heptadecapeptide termed orphanin FQ (or nociceptin), which is structurally similar to the endogenous opioid peptide dynorphin, seems to be the endogenous ligand for ORL-1 (which has been renamed the N/OFQ receptor [NOP]). Nociceptin does not act at any of the three cloned opioid receptors, however, and the nociceptin–NOP ligand–receptor complex may instead function to facilitate pain at some sites.
Another heptadecapeptide, termed nocistatin, is also derived from the same gene that gives rise to nociceptin. Nocistatin is reported to “block” the effects of nociceptin, but nocistatin does not displace nociceptin from its receptor (ORL-1),26 indicating that the opposing effects of nocistatin are produced at a yet to be identified receptor.49 It is believed now that nocistatin exerts an inhibitory effect through a presynaptic Gi/Go-coupled receptor not related to either the opioid receptors or the NOP receptor.5
The μ receptor is the site at which all the currently available pure agonists act to produce analgesia. Morphine and other similar pure agonists have greatest affinity for μ receptors in the CNS and peripheral nervous system and lower affinity for δ and κ receptors. Among the endogenous opioid peptides, the endomorphins have the highest affinity for the μ receptor; β-endorphin and the enkephalins also exert some of their effects at μ receptors. Two subtypes of μ receptor have been characterized by pharmacologic means: μ1, which is associated with supraspinal analgesia, and μ2, which subserves spinal analgesia, respiratory depression, and gastrointestinal actions. Pharmacologic studies have also implied the existence of two subtypes of δ receptor, at which the endogenous enkephalins are considered to be the prototypic agonists, although the enkephalins also interact with μ receptors. The δ receptor is also involved in analgesia, mainly through spinal mechanisms, and in causing, along with μ receptors, opioid reinforcement (see Chapter 51).
Dynorphins, representing the third class of endogenous opioid peptides, have been proposed to be ligands for the κ receptor, of which three subtypes have been characterized pharmacologically. Two κ receptors mediate spinal (κ1) and supraspinal (κ3) analgesia and are principally responsible for the analgesic effects of the mixed agonist-antagonist group of opioid analgesics currently available. Although the endomorphins selectively bind to the μ opioid receptor,48 other endogenous opioids and clinically available drugs do not bind selectively to specific opioid receptors. Although morphine and other pure agonists preferentially bind at μ receptors, they can produce effects at δ and κ opioid receptors as well, particularly as the dosage is increased.
Despite pharmacologic evidence for the existence of multiple subtypes of opioid receptors, only one of each opioid receptor has been cloned. There is no evidence at present for the existence of genes other than those that give rise to the already cloned μ, δ, and κ receptors. It is possible that not all opioid receptor genes have been cloned, but molecular mechanisms and other factors most likely explain the pharmacologic diversity of effects produced by various ligands at opioid receptors. Receptor isoforms may be produced by alternative splicing in the coding regions of the three cloned opioid receptors.7 Variant mRNAs have been reported for all three opioid receptors, although their expression is typically very low, and it is not clear that splice variants can be distinguished pharmacologically. It seems more likely that other mechanisms (e.g., post-translational regulation, variable activation of receptors by different ligands, receptor dimerization, intracellular interactions with proteins associated with different effectors) explain the pharmacologic diversity of opioid receptor subtypes. Many G protein–coupled receptors exist as dimers (two receptors linked), and heterodimerization of δ and κ receptors could result in a complex pharmacologic profile.
Physiologic functions
Considerable attention has been given to the notion that endogenous opioid peptides tonically modulate nociception (pain). One would expect occupation of opioid receptors by naloxone to prevent any action by endogenous opioid peptides and lower the response threshold for pain if endogenous opioids tonically modulated nociception. A number of studies have shown that the administration of naloxone to normal human volunteers does not affect their resting pain thresholds or responses to experimental pain stimuli. In other situations, naloxone has been reported to attenuate the analgesic effects of acupuncture and of placebo administration after minor oral surgery and to be hyperalgesic in humans after major surgery.31 From these and other findings,8 it seems that endogenous antinociceptive opioid systems are normally quiescent but can become physiologically active and affect pain processing when significant injury or stress is present. It seems that endogenous opioid activity can be enhanced by the anticipation of pain relief, producing naloxone-reversible placebo effects that diminish the perception of pain (i.e., elicit analgesia).6
Sites and mechanism of action
Among the many effects of opioids, analgesia has been most thoroughly studied. We now know that opioids acting at opioid receptors in the periphery and at spinal and supraspinal sites can produce clinically effective analgesia. Knowledge of the central sites of opioid action came first; documentation of direct peripheral analgesic actions of opioids is relatively recent.38
Early investigators attempted to determine the central locus of morphine’s analgesic action by administering morphine directly into selected brain sites. These studies succeeded in identifying an area of the brainstem surrounding the cerebral aqueduct as a site important to morphine analgesia. Corollary studies found that electrical stimulation of this same area in the midbrain in animals and humans produced a potent, long-lasting analgesia partially mediated by endogenous opioids.11 Morphine and other opioid agonists acting at this site produce analgesia by engaging a descending system of pain inhibition.8
As illustrated in Figure 20-3, information about pain from nociceptors—that is, peripheral receptors in skin, muscle, joints, and viscera that respond to pain-producing stimuli—can be influenced at the first central synapse (spinal or medullary dorsal horn) by this descending system of pain inhibition. It is important to appreciate two features of this pain-modulating system: the descending pathway from the midbrain is indirect (there is a synaptic relay in the medulla or the dorsolateral pons or both), and although engaged by an opioid acting at opioid receptors in the midbrain, the neurotransmitters in the spinal cord or medullary dorsal horn that ultimately inhibit pain transmission are nonopioids such as 5-hydroxytryptamine (5-HT, serotonin) and norepinephrine.8 Engagement of the descending pain inhibitory circuit is also responsible for placebo-induced analgesia.
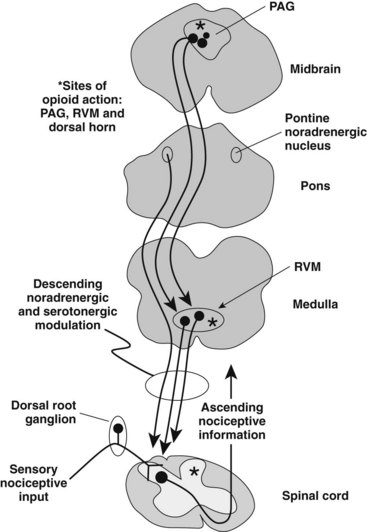
A second means by which morphine and other opioid agonists modulate pain is by acting at opioid receptors in the brain at sites not associated with activation of the descending pain inhibitory system. Actions of opioids at these brain sites do not affect the response threshold to a painful stimulus but rather influence the interpretation of and emotional reaction to the stimulus.31 This so-called motivational-affective component of the response to pain is discussed later in this chapter.
A final means by which morphine and other opioid agonists produce analgesia is by acting at opioid receptors located on the peripheral and central terminals of nociceptors. Opioid receptors are located on the terminals of these specialized nerve fibers, typically Aδ and C fibers, which connect the periphery directly with the CNS. Analgesic effects of opioids can be produced by direct administration of an opioid into a joint or, more commonly, the epidural or intrathecal space.46
Opioids can act at several sites, each of which can contribute independently to the analgesia produced. When given systemically, opioids have access to all potential sites of action, and the analgesia produced is likely a product of peripheral, spinal, and supraspinal interactions with opioid receptors. These mechanisms are likely to be synergistic resulting in increased opioid potency and, consequently, reduced toxicity. This knowledge can be used to improve pain control and limit the incidence or severity of undesirable opioid effects. Because pain modulation descending from the brainstem is mediated in the spinal cord by norepinephrine and 5-HT, the direct effects of an opioid given into the epidural space can be enhanced by epidural administration of an α-adrenoceptor agonist such as clonidine.24
Tricyclic antidepressants, which block the reuptake of norepinephrine and 5-HT, are also effective adjuvants (although antidepressants such as amitriptyline also possess analgesic efficacy unrelated to their effects on monoamine reuptake). This strategy allows reduction of the dose of opioid without compromising the analgesia produced. Two drugs, tramadol and tapentadol, produce analgesic actions by engaging multiple mechanisms, including activity at the opioid receptors and inhibition of reuptake of norepinephrine or serotonin or both. By requiring less opioid for adequate pain control, undesirable opioid effects, such as urinary retention, respiratory depression, sedation, and development of analgesic tolerance, can be reduced. It has also been documented that administration of very low doses of morphine directly into the knee joint after arthroscopic knee surgery can control postoperative pain.38
The mechanisms by which opioids produce their effects are best established for direct actions at opioid receptors located on neurons. Actions commonly produced at all three opioid receptors include inhibition of adenylyl cyclase, inhibition of Ca++ conductance, activation of K+ conductance, and inhibition of neurotransmitter release (see Figure 20-2). Acute inhibition of adenylyl cyclase by an opioid leads to a decrease in intracellular cyclic 3′,5′-adenosine monophosphate, decreasing an inward, nonselective cation current and decreasing cell excitability. All three opioid receptors also activate a G protein inwardly rectifying K+ conductance and inhibit voltage-activated Ca++ currents, both events mediated by G protein β-γ subunits. Because Ca++ influx is required for the stimulus-secretion coupling of neurotransmitter release, opioids decrease the release of excitatory neurotransmitters, such as glutamate, substance P, and calcitonin gene–related peptide, from nociceptor terminals and attenuate the transmission of nociceptive information at the first central synapse. Because opioids activate this G protein–rectifying K+ conductance, they produce a relative hyperpolarization of neurons, making them more difficult to excite.
Morphine
The structure of morphine is shown in Figure 20-4. Morphine is the prototypic opioid analgesic (pure agonist) and the one about which most is known. Morphine is widely used for pain control and can be given by virtually any route of administration. All opioid analgesics share with morphine the ability to produce analgesia, respiratory depression, constipation, gastrointestinal spasm, and physical dependence; none has yet been shown to be significantly different from or superior to morphine regarding its important pharmacologic features. The incidence of untoward effects (e.g., respiratory depression) and the intensity of action of pure agonists are qualitatively similar and differ little when compared at doses that produce equivalent analgesia. Consequently, morphine is discussed in greater detail than other opioid analgesics, and what is stated for morphine applies in general to other pure agonists. Significant differences that exist between morphine and other opioids are mentioned as each individual agent is discussed.
Central pharmacologic effects
Analgesia
As already discussed, the sites of opioid-produced analgesia include the periphery and spinal and supraspinal brain areas. It is generally accepted that opioid-induced analgesia involves the sensory-discriminative and motivational-affective components of pain. The sensory-discriminative component of pain is associated with identification and localization of the source of pain, whereas the motivational-affective component of pain is related to one’s reaction to pain.33 Pain is not a simple sensation associated with a single pain pathway from the periphery to the cortex, but rather, it is a complex experience that can be influenced by the environment in which the pain arises: prior experience and expectation; attention, anxiety, mood, and stress levels; and other societal, emotional, and cognitive contributions. The nociceptive component of pain may not be as much affected by opioid analgesics as is the reaction to pain. A common report from patients after receiving an opioid for relief of pain is that the pain is still present, but that it is not discomforting. Clinical impressions and patients’ reports suggest a prominent action by opioid analgesics on the motivational-affective component of pain, presumably resulting from opioid actions at opioid receptors within the limbic system of the brain.
An additional significant feature of opioid analgesics is that they are generally more effective against continuous, dull, aching pain than sharp, intermittent pain. It is also known that sensitivity to pain and the ability to clear morphine decrease with age, whereas the elimination half-life of morphine increases with age; the pain relief provided by morphine typically increases with age.14,28
Respiratory depression
Morphine and its congeners depress respiration in a dose-related fashion. Respiratory depression represents the principal undesirable, potentially life-threatening effect of opioids as a group. Opioids are capable of depressing the tidal volume and the rate of respiration. In humans, morphine decreases the response of brainstem respiratory centers to the carbon dioxide tension of the blood. It also significantly depresses pontine and medullary centers that regulate respiratory frequency.2 Irregular rhythms and periodic breathing are common after toxic doses of morphine or its congeners, and the normal respiratory rate of 16 to 18 breaths/min may be reduced to 3 to 4 breaths/min. All currently available opioid analgesics are capable of depressing respiration in a manner similar to that of morphine when administered in doses that produce equal analgesia.
Peripheral pharmacologic effects
Gastrointestinal tract
The use of opium for relief of diarrhea and dysentery antedated by centuries the use of opium for relief of pain. Opioids exert significant effects on smooth muscle all along the gastrointestinal tract. The overall action of morphine and its congeners is constipating, an effect that is useful therapeutically. Opioid analgesics, acting principally at opioid receptors in the gastrointestinal tract but also at opioid receptors in the CNS,41 increase smooth muscle tone and decrease propulsive motility throughout the gastrointestinal tract. In the large intestine, muscle spasms can result from the marked increase in muscle tone and nonpropulsive muscle contractions. Spasm of the smooth muscle of the biliary tract, which can be very painful, can also occur after the administration of therapeutic doses of morphine and related drugs.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


