Wound Healing
The capacity for self-repair is crucial for the survival of any organism, because without it the organism would likely perish after minimal injury. A wound is a disruption in the normal anatomic structure and function of tissue and is accompanied by cellular damage. Wound healing is an intricately coordinated series of processes that involve cellular and subcellular responses to tissue injury, leading to the release of cytokines and growth factors, cell activation, and resultant tissue regeneration.1,2 The large variation in repair capacity of different tissue types is intriguing. For example, hepatic tissue has a high capacity to regenerate, whereas nerve tissue has an exceptionally low repair potential, given its inability to replicate. A solid understanding of the repair process is essential for optimizing patients’ perioperative healing and is the basis for minimizing iatrogenic injury. It is especially important for surgeons treating maxillofacial injuries to possess a thorough knowledge of the wound-healing process, because nowhere else in the body are the effects of poor healing more noticeable and potentially disfiguring. To optimize the restoration of function and esthetic harmony after facial trauma, the surgeon must also be cognizant of patient-specific comorbidities and understand how health status influences the healing process.3,4
The challenge to optimize healing has placed wound physiology at the forefront of clinical and laboratory research.5 The understanding of the remarkable cascade of events involved in wound repair and healing is advancing exponentially with the ongoing discoveries of the roles of growth factors and signaling pathways. There is growing interest in stem cell research, regenerative medicine applications, and bioactive wound healing products.
Wounds are classified as acute or chronic. Acute wounds have surgical, traumatic, pathologic, or ischemic causes. Surgical wounds, intentionally created in the operating room environment, vary in their degree of contamination, depending on their anatomic location and presence of local microbial flora. Subsequent healing is affected by the level of cleanliness or contamination of the wound. Traumatic injuries caused by blunt or penetrating trauma result in tissue laceration, abrasion, or even tissue avulsion. Other mechanisms of traumatic injury include tissue exposure to extremes of temperature, radiation, or caustic chemicals that cause injury by altering tissue pH, denaturing proteins, and causing local ischemia.1,2 Pathologic processes such as neoplasms and nonhealing ulcers also cause tissue disruption. Skin breakdown or ulceration secondary to ischemia is related to impaired blood flow to an area by vascular occlusion, compression, stasis or pressure. Traumatic handling of tissues during treatment, including crush injuries and desiccation, can add further insult to the initial injury.
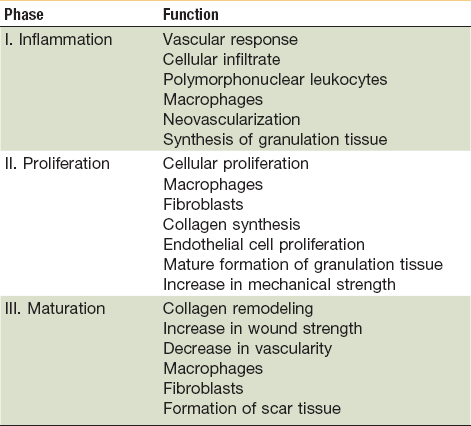
In the state of health, wound healing occurs in three distinct but overlapping phases—inflammation, proliferation, and remodeling (Table 2-1).1–4,6 When the tissue’s normal healing process experiences a disruption or delay, a chronic wound forms. Delays usually arrest healing at the inflammatory phase and result in excessive collagen deposition and scarring. Local factors impairing wound healing include the presence of foreign bodies or necrotic tissue within the wound, a high microbial burden, ischemia secondary to venous or arterial insufficiency, and tissue hypoxia secondary to radiation fibrosis. Some systemic factors that reduce healing capacity include aging, malnutrition, vitamin deficiencies, diabetes, immunocompromised states, atherosclerosis, peripheral artery disease, collagen vascular disease, and chemotherapy.7
General Concepts of Wound Healing
Normal Soft Tissue Healing (Repair)
Normal wound healing results in tissue regeneration and takes place in three separate but overlapping phases—inflammation, proliferation and remodeling (Fig. 2-1).1–4,8–10
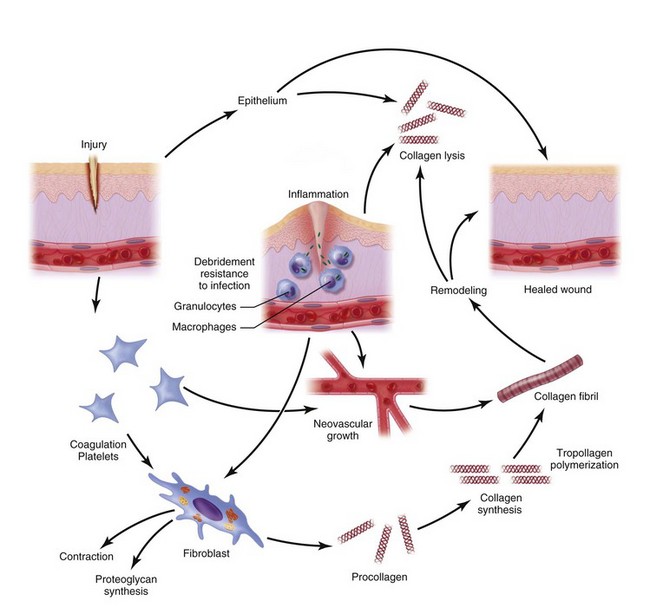
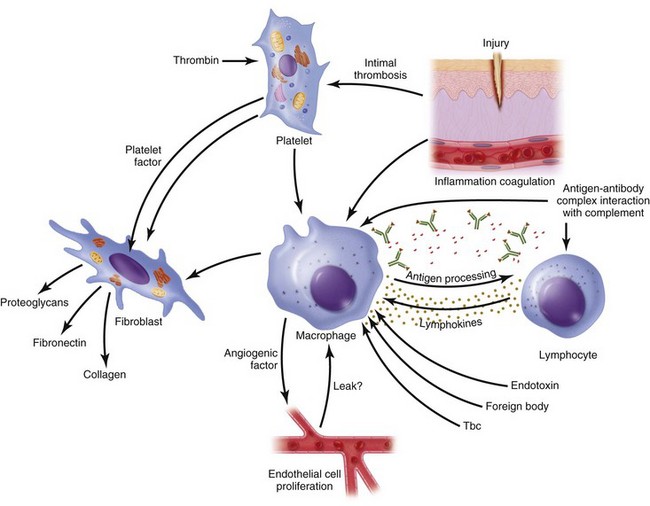
The inflammatory phase of wound healing begins at the time of injury and lasts for 3 to 5 days. Vasoconstriction initiates the process, because catecholamines and prostaglandins (epinephrine and thromboxane) cause small blood vessels to constrict for initial hemostasis. Hemostasis via vasoconstriction is short-lived but is soon followed by the formation of a blood clot. The clotting cascade, prompted by vessel disruption, is initiated by platelets. Platelets, fragments of megakaryocytes that circulate in the blood for 9 to 11 days, contain glycogen, dense granules, and alpha granules (Fig. 2-2). When platelets adhere to the exposed subendothelial collagen of injured vessels with the aid of von Willebrand factor, they degranulate, releasing adenosine triphosphate, serotonin, prostaglandins, and thromboxane A2.3,4 Serotonin, prostaglandins, and kinins increase vascular permeability. Platelets also release interleukins (ILs) and growth factors (transforming growth factor β [TGF-β], platelet-derived growth factor [PGDF] and vascular endothelial growth factor [VEGF]), which further potentiate platelet activation and aggregation. Insulin-like growth factor 1(IGF-1), TGF-α, TGF-β, and PGDF attract leukocytes and fibroblasts to the wound. These factors also serve as chemoattractants when released into the bloodstream, recruiting neutrophils and monocytes to the site of injury. As platelets continue to aggregate, they form a platelet plug within the vessel. The complement, kinin, plasminogen, and clotting cascades are activated. The clotting cascade results in the deposition of fibrin, a key component that strengthens the platelet plug and acts as a scaffold for wound healing.
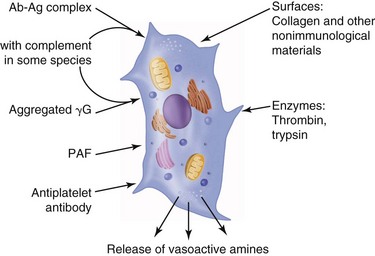
Vasodilation and inflammation follow hemostasis. This process is mediated by a variety of cytokines and factors, including histamine from mast cells (Fig. 2-3; Table 2-2), prostaglandins PGI2 and PGE2, prostacyclin, platelet-activating factor (PAF), bradykinin, leukotrienes, and nitric oxide. These chemicals cause leukocytes and plasma proteins to permeate into the wound. The endothelial cells of the small venules and capillaries transiently change shape and become rounded, creating gaps within the vessel wall that allow for leaking of plasma and fibrinogen.3,4 Leukocytes migrate to the extravascular space via diapedesis (Fig. 2-4). Histamine, produced by mast cells and basophils, increases vascular permeability by inducing endothelial cell contraction and exposing the endothelial basement membrane. Consequently, histamine receptor blockers can prevent early changes in vascular permeability. Increased vascular permeability leads to the clinical findings of calor, rubor, dolor and tumor.1 Wound heat and redness are a result of vasodilation and the presence of blood-borne cells. Pain, a nerve response caused by vasoactive amines or pressure from edema, protects the site from further tissue injury. Finally, wound swelling is caused by increased volume at the site of injury, secondary to edema from vascular permeability.
TABLE 2-2
Chemical Mediators Derived from the Mast Cell
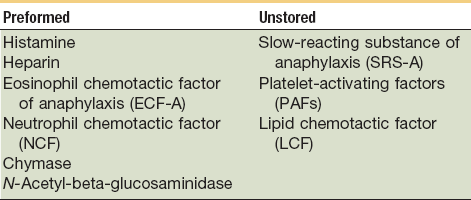
From Yurt RW: Role of the mast cell in trauma. In Dineen P, Hildrick-Smith G, editors: The surgical wound, Philadelphia, 1981, Lea & Febiger, pp 37–62.
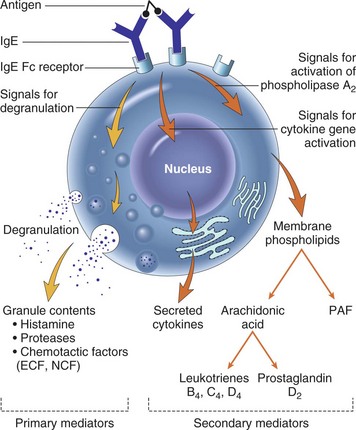
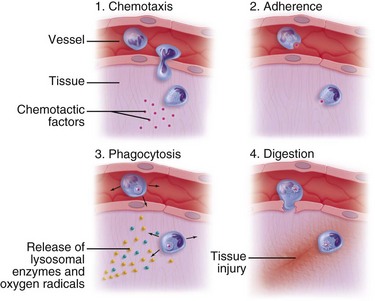
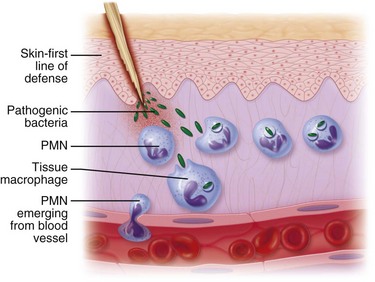
Some of the first cells to be recruited to the site of injury are neutrophils, which move across the endothelium via diapedesis within minutes of injury (Fig. 2-5). The neutrophil concentration within the wound peaks by 24 hours. They autolyse to release intracellular contents into the wound, including lysosomic proteases for degradation of nonviable tissue, debris, and bacteria (Table 2-3). Collagenases, elastases, cathepsin, and bactericidal cationic proteins are also released from neutrophil granules. Collagenases and cathepsin G activate complement and aid in the conversion of kininogens to kinins. Monocytes follow and concentrate at the site of inflammation within 2 to 3 days. These cells are paramount in directing wound healing because they transform into macrophages that continue wound débridement via the secretion of hydrolytic enzymes into the extracellular space. Collagenases, elastases, and cathepsins catalyze the conversion of plasminogen to plasmin to begin clot breakdown. Macrophages engage in microbial phagocytosis, a process enhanced by infiltrating opsonins. Macrophages release additional chemoattractant substances and growth factors that recruit fibroblasts and stimulate collagen production. Growth factors necessary for the formation of granulation tissue include TGF-α, TGF-β1, PDGF, epidermal growth factor (EGF), fibroblast growth factor (FGF), IGFs, TNF-α and IL-1. Neutrophils and macrophages continue to release cytokines that will initiate the proliferative phase of healing.
TABLE 2-3
Injurious Constituents of Neutrophils
| Constituent | Activity |
| Collagenase, elastase, and cathepsin A | Hydrolysis of basement membranes, internal elastic laminae, cartilage and other connective tissue; generation of C5 fragments, angiotensin II |
| Basic proteins (three) | Increased vascular permeability |
| Basic protein (one) | Activation of mast cells, release of vasoactive amines |
| Leukotrienes C4 and D4 | Increased vascular permeability, contraction of smooth muscle |
| Kininogenase | Hydrolysis of kininogen with release of vasoactive kinin |
| Procoagulant activity | Generation of fibrin, activation of platelets |
| Platelet activation factor (PAF) | Activation of platelets, increased vascular permeability, contraction of smooth muscles, activation of neutrophils |
| Leukotriene B4 | Attraction of leukocytes |
| Lysosomal enzymes | Digestion of tissue constituents |
| Oxygen radicals | Damage to cells |
From Bellanti JA: Immunology, ed 3, Philadelphia, 1985, WB Saunders, p 258.
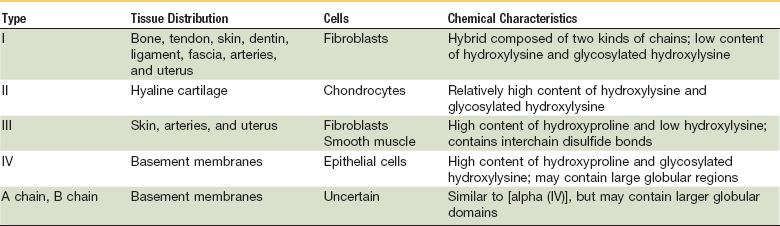
Continuing after the inflammatory phase, the proliferative phase (also known as the fibroblastic stage) is established by the fourth or fifth day and lasts 2 to 3 weeks. It is characterized by the ingrowth and proliferation of granulation tissue within the wound. Granulation tissue, a loose connective tissue matrix formed by collagen-secreting fibroblasts, supports neovasculature and inflammatory cells (Fig. 2-6). Fibroplasia, angiogenesis, and subsequent epithelialization further typify the proliferative phase. Responding to the release of PDGF and TGF-β, fibroblasts arrive at the wound on the third day and peak in concentration within 1 week. They actively produce proteoglycans and collagen, with force, stress, strain, and motion directing the collagen and proteoglycan alignment. Fibroblasts synthesize mostly type III collagen for approximately 3 weeks, until equilibrium is reached between the production and breakdown of collagen (Table 2-4). Budding vessels closely follow fibroblast activity. Neovascularization is enhanced by local factors such as hypoxia, elevated tissue lactate levels, and cytokines, such as FGF, VEGF, and PDGF.1–4 Angiogenesis is crucial because new vasculature is required for the influx of oxygen and nutrients and the removal of metabolic waste products. The granulation tissue contains inflammatory cells and fibroblasts in a matrix of collagen and new vasculature. Epithelialization is promoted by EGF, TGF-α, and keratinocyte growth factor, and is itself composed of three phases—epithelial migration, proliferation, and differentiation. The dermal layer is reepithelialized and the contractile forces exerted by fibroblasts and myofibroblasts aid in reapproximating wound margins. Skin-grafting open soft tissue wounds can limit the amount of granulation tissue produced, thereby reducing scarring and tissue contracture. In the head and neck region, reepithelialization occurs faster in mucosa than in skin. Mucosal reepithelialization occurs over a moist surface whereas reepithelialization in skin occurs under a dry scab. The mechanical strength of the wound increases through the proliferative phase.
TABLE 2-4
From Prockop DJ: Collagen biochemistry and the design of agents to inhibit excessive accumulation of collagen during wound repair. In Dineen P, Hildrick-Smith G, editors: The surgical wound. Philadelphia, 1981, Lea & Febiger, p 97.
Tissue remodeling finalizes the wound healing process. Also known as maturation, the remodeling phase is characterized by an increase in wound tensile strength related to increased collagen production and breakdown. The remodeling process begins after the third week and usually lasts 6 to 12 months. Initial scar tissue becomes stronger as type I collagen replaces type III collagen. The augmented collagen deposition and subsequent collagen cross-linking increase the scar’s tensile strength to 75% to 80% of the preinjury tensile strength. Tensile strength is defined as the load per cross-sectional area that can be supported by a wound; the increase in tensile strength is proportional to the rate of collagen synthesis. Over time, a decrease in the number of fibroblasts and macrophages is seen, with scar vascularity also decreasing as tissue proliferation declines.1,2 These changes are clinically correlated to a less erythematous, flatter, softer scar.
Abnormal Soft Tissue Healing (Repair): Keloids and Hypertrophic Scars
Keloids and hypertrophic scars are aberrant forms of wound healing that result in proliferative scarring.11 Although clinically similar, they differ in their formative timeline and boundaries. Keloids are benign growths of fibrous tissue that grow beyond wound boundaries. They more commonly occur over the sternum, ear lobes, back, trunk, and extremities. Keloids are firm and rubbery and can be erythematous, painful, or pruritic. They develop months after trauma, piercing, or surgical incisions and are caused by an overproduction of connective tissue, likely secondary to altered apoptosis or hyperproliferation of keloidal fibroblasts. Genetic causes have also been implicated in keloid formation. Keloids occur more commonly in certain ethnic populations. The incidence of keloid formation is correlated with increased skin pigmentation. Keloids rarely improve without treatment and a variety of treatment modalities have been described. First-line treatment is intralesional injection of corticosteroids into the scar to reduce fibroblastic production of collagen and extracellular matrix proteins. Localized pressure therapy, interferon, or fluorouracil can be used in combination with intralesional corticosteroid injections. Keloids can also be surgically excised or treated with radiation, cryosurgery, or topical imiquimod. The use of calcineurin inhibitors is currently under investigation. Nevertheless, regardless of treatment modality, keloids commonly recur to some degree after treatment.12
Hypertrophic scars have a similar appearance to keloids but differ from keloids in that they do not extend beyond the margins of the original wound. They also appear shortly after injury and may recede over time. Hypertrophic scars, characterized by prolonged inflammation and collagen deposition, are red, firm, and elevated. Hypertrophic scars are also treated with intralesional corticosteroid injections and are less likely than keloids to recur after treatment.13
Wound Repair in Other Tissues of the Head and Neck
Normal Bone Healing (Repair)
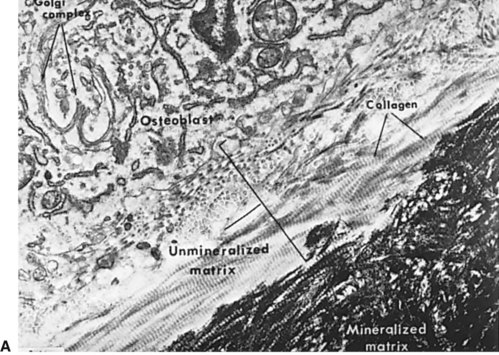
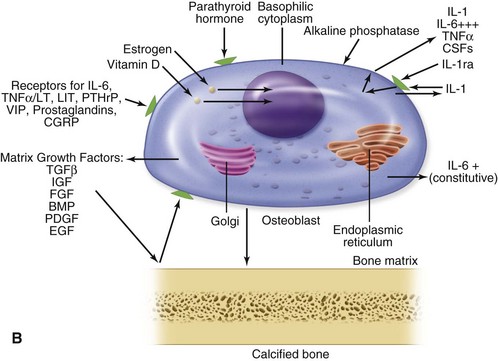
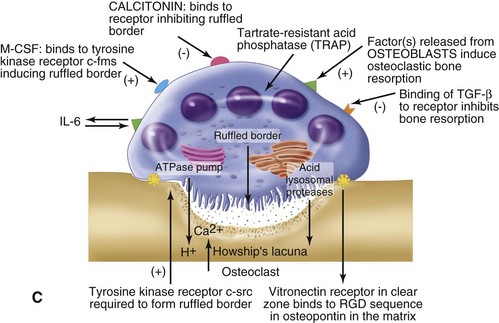
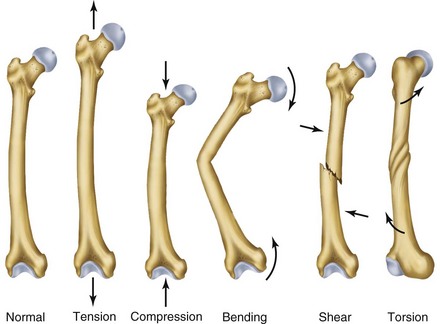
Normal bone healing parallels soft tissue healing. Both tissue types undergo three phases of wound healing—inflammation, proliferation, and remodeling.3,10,14,15 However, bone healing also undergoes calcification. As with soft tissue repair, the inflammatory stage in bone healing begins immediately after the injury and lasts up to 5 days. Inflammation is stimulated by vessel injury in the haversian canals and periosteum and by the presence of bony debris or necrotic material in the fracture site. Vasoconstriction allows a blood clot to form, and inflammatory cells phagocytize debris and bacteria. A hematoma is formed within the fractured bone. The necrotic bone edges are resorbed. The proliferative fibroplastic stage follows. Pleuripotential mesenchymal cells and fibroblasts enter the site of injury to lay down fibrous tissue, cartilage, and immature bone fibers (Table 2-5). These components permit the wound to gain some strength over the 2 to 3 weeks following injury. Granulation tissue forms as a matrix of fibrin, collagen, and neovasculature is laid down. If the fracture segments are not precisely reduced to the preinjury anatomic position, or if bone is avulsed, leaving a residual space between the two bony segments, the fracture will heal by secondary intention. Greater collagen deposition is then required to bridge the gap, resulting in callus formation at the surface and within the fractured bone. The soft cartilage callus calcifies into woven bone as osteoblast and osteoclast concentrations increase within the fracture site (Fig. 2-7). Osteoblasts continue to deposit osteoid on spicules of calcified cartilage and this osteoid is later calcified to immature bone (Fig. 2-8). The callus, much like a rudimentary splint, offers the fractured bone some minor stability against bending and torsion (Fig. 2-9). However, immobilization is required for healing to proceed; otherwise, a fibrous union will result.
TABLE 2-5
Polypeptide Growth Factors Involved in Bone Healing
| Stage of Bone Healing | Growth Factor | Function(s) |
| I. Vascular | Plasma fibronectin | Anchors cells in the ground substance; ingrowth required for collagen formation |
| Endothelial cell–derived growth factor | Mitogen | |
| II. Callus | Platelet-derived growth factor | Mitogen—fibroblasts, bone cell formation; activates monocytes; promotes bone resorption |
| Epidermal cell growth factor | Mitogen—cartilage, bone; inhibits type I bone collagen synthesis | |
| Fibroblast growth factor | Mitogen—fibroblasts, chondrocytes | |
| Insulin-like growth factor | Chondrocyte proliferation; chondrocyte proteoglycan synthesis | |
| Nerve growth factor | Mitogen | |
| III. Bone formation, remodeling phase | Epidermal growth factor | Promotes bone resorption |
| Fibroblast growth factor | Promotes bone resorption in high doses | |
| Insulin | Synergistic effect with bone growth factors | |
| Interleukins (monocyte products) | IL-1: Fibroblast proliferation, collagenase production, prostaglandin production IL-2: T cell growth factor, stimulation of bone resorption by osteoclastic activation factor (OAF) production |
From Simmons DJ: Fracture healing perspectives. Clin Orthop Relat Res 200:100, 1985.
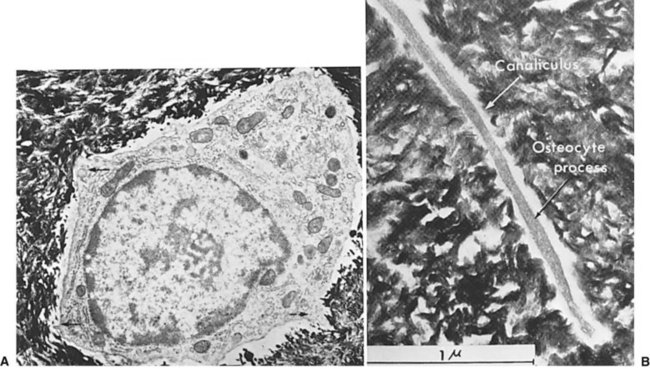
In areas adjacent to endosteum, where the vascular supply and osteoprogenitor cells abound, no intermediate fibrocartilage is seen. Instead, the endosteal osteoblasts form a direct bony callus. Similarly, no cartilaginous callus is formed when minimally displaced fractures heal under immobilization or when acute fractures are anatomically reduced without a gap between the segments and immobilized with rigid fixation. This type of fracture healing is known as primary intention (Fig. 2-10).
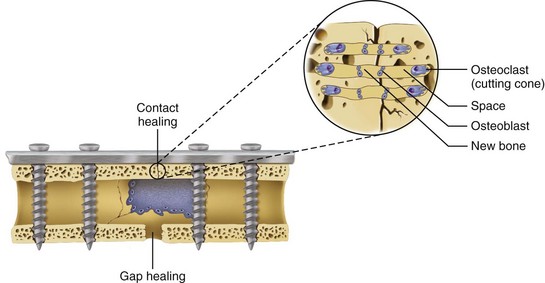
As long as the fracture site remains immobilized, maintains an adequate blood supply, and remains free of infection, the remodeling stage will complete the fracture healing process,. The callus completely ossifies as osteoclasts gradually resorb the immature bone and it becomes remodeled into lamellar bone. The gradual osteoclastic resorption of immature woven bone with osteoblastic bone formation and maturation to lamellar bone is known as creeping substitution.3,10,14
Dental extraction sites heal by secondary intention. The socket first fills with blood that quickly coagulates within the first 24 hours to form a blood clot that seals the alveolar socket from the oral cavity. Inflammation ensues soon after the injury of dental extraction. During the first week of healing, leukocytes débride the extraction site, phagocytose bacteria, and débride bony fragments. Osteoclasts resorb the marginal bone along the extraction socket (Fig. 2-11). Meanwhile, epithelial cells begin migrating along the socket wall to reepithelialize the socket surface. During the second week of healing, granulation tissue is generated while osteoid is deposited by osteoblasts to form woven bone. By the fourth week of healing, full epithelialization is achieved. The alveolar bone is remodeled over 4 to 6 months. During this time, the alveolar cortical bone and trabecular woven bone are resorbed and replaced by lamellar bone.10,14
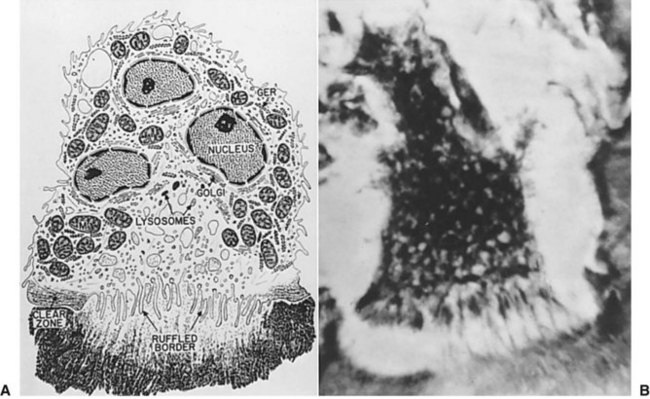
Complications in Bone Healing
Repeated trauma to an area undergoing bone healing can result in malunion or nonunion of a bone fracture (Fig. 2-12). A malunion results when the bony fracture segments heal in an incorrect or nonanatomic position, which can lead to a deformity.16 For fractures of the jaws, malunion will create a malocclusion. A bony nonunion results when the fracture segments do not form bone to bone contact, but instead remain bridged by fibrous tissue. In the maxillofacial region, bony nonunion is most commonly seen in inadequately treated mandible fractures.17 The mandible is a bone with high functional forces and repeated trauma can occur if the patient masticates before bone healing is complete.18 Tobacco use and excessive alcohol intake increase the risk of nonunion.19 Nicotine impairs bone healing by preventing vascular ingrowth and diminishing osteoblast function during the proliferative stage.20,21 Use of certain anti-inflammatory or cytotoxic medications (e.g., nonsteroidal anti-inflammatory drugs [NSAIDs], glucocorticoids, chemotherapeutic agents, fluoroquinolone antibiotics) during the inflammatory phase of bone healing has also been implicated in an increased risk of nonunion,22 but the actual clinical significance may be negligible. A nonunion of the mandible can lead to severe pain, instability of the mandibular fracture segments, and malocclusion. A nonunion requires open surgical treatment, with removal of the fibrous callu/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


