Recognition and Management of Shock
Treatment of shock varies depending on the type of shock. There are four major categories of shock—hypovolemic shock (hemorrhagic versus nonhemorrhagic), cardiogenic shock, obstructive shock, and distributive shock (septic, anaphylactic and neurogenic).1 Each category of shock may present with variable clinical signs and symptoms and requires different treatments. For example, aggressive volume replacement would be appropriate in a patient who is in hypovolemic shock, but may be detrimental in a patient with cardiogenic shock. Therefore, one must not only recognize shock but also be able to categorize it and implement the appropriate treatment based on the available clinical data. This chapter will give an overview of the cellular and systemic response to shock and then focus on its recognition, categorization, and current treatment philosophies.
Cellular Changes
Organ hypoperfusion creates a hypoxic environment that initiates cellular changes resulting in shock. At the cellular level, shock occurs when the delivery of oxygen is inadequate for cells to metabolize glucose for energy needed to run the cellular machinery. Cells depend on oxygen to carry out aerobic respiration via the mitochondria. Mitochondria are the power plants of cells that supply 36 adenosine triphosphate (ATP) molecules through the oxidation of each molecule of glucose. Without an adequate supply of oxygen, mitochondria switch to anaerobic respiration, yielding only two ATP molecules and lactate per glucose molecule. Lactate lowers the tissue pH and is released into the systemic system, becoming a useful marker for tissue hypoxia and acidosis. Not only is lactate toxic to cells, but a lack of ATP generation leads to free radical formation and damage to vital cell functions, most notably ATP-dependent ion channels that maintain normal membrane potentials. When ATP-dependent Na+, K+, and Ca2+ transport are disrupted, cellular membranes lose their electrical gradient, leading to intracellular bleb formation, cellular edema, and cell lysis. The tipping point for irreversible cell damage is not entirely understood but is thought to be a result of uncontrolled release of intracellular Ca2+ stores. Intracellular bleb formation and mitochondrial swelling are signs of irreversible cellular dysfunction that leads to cell lysis. When intracellular enzymes and cellular material are released into the surrounding tissue and circulation, further inflammation, edema, and tissue damage occur.2,3
Endothelium normally produces nitric oxide but when damaged by inflammation, nitric oxide synthase is overexpressed, producing toxic levels of nitric oxide and other oxygen-derived free radicals that are formed when tissue are reperfused with oxygen. This reperfusion injury creates free radicals and further activation of inflammatory mediators, notably interleukin-8 (IL-8), IL-1, and tumor necrosis factor (TNF). The combination of ATP depletion, tissue edema, pH changes, free radical formation, and inflammatory mediator formation leads to a complex vicious cycle of cell death and irreversible organ damage.3
Systemic Response
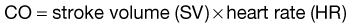
or
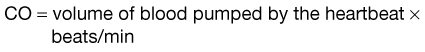
Not only does reducing the renal excretion of Na+ and water increase the intravascular volume, but the body is able to regulate changes in transcapillary fluid movement to improve intravascular volume. This occurs with increases in capillary oncotic pressure and a decrease in hydrostatic pressure. When arterioles constrict, blood flow and hydrostatic pressure through capillary beds is greatly reduced, causing a net shift of fluid into the vascular space. Also, larger molecules are unable to cross capillary fenestrations creating an oncotic pressure that drives extravascular fluid into capillaries. This oncotic pressure is magnified when catecholamines stimulate glycogenolysis and gluconeogenesis, causing an increase in the blood glucose concentration. These compensatory mechanisms are limited, and definitive treatment must be initiated quickly and will vary depending on the type and severity of shock.4
Major Categories of Shock
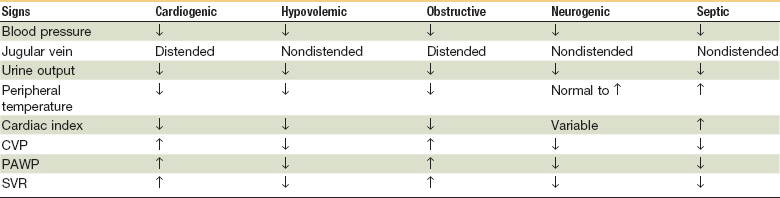
Hypovolemic Shock
The most commonly observed form of shock, hypovolemic shock, results from a rapid loss of intravascular volume; this can be further subdivided into hemorrhagic and nonhemorrhagic types (Box 7-1). Trauma and/or GI bleeding are the most common sources of hemorrhagic shock, and exsanguination is responsible for 80% of deaths in the operating room and almost 50% of deaths in the first 24 hours after injury.5 Retroperitoneal accumulation is always a diagnostic challenge and the presence or amount of blood loss is frequently underestimated. Nonhemorrhagic shock results from increased noncompensated fluid loss (e.g., dehydration, vomiting, diarrhea, polyuria, fluid redistribution), as seen in burns and anaphylaxis.
Class II hemorrhage is a blood volume loss between 15% and 30%. Clinical symptoms include tachycardia, tachypnea, and a decrease in pulse pressure. These patients are normally stabilized and usually respond to expansion of the intravascular volume with crystalloids, but some patients may require blood transfusion.1 Patients with class II blood loss can usually sustain normal pressures while supine but may be orthostatically dependent.6
Class IV hemorrhage is a blood volume loss of more than 40% and is an imminent life-threatening situation. Symptoms include a significant depression in systolic blood pressure and a very narrow pulse pressure. Urinary output is negligible and mental status is depressed.1 These patients exhibit an impaired level of consciousness and will experience a precipitous death if intravascular volume is not quickly restored (Table 7-2).
TABLE 7-2
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


