CHAPTER 17 Principles of General Anesthesia
HISTORY
Finally, the use of nitrous oxide by dentists has exhibited a cyclic pattern of popularity every 25 to 30 years since Wells first used it. Nitrous oxide is currently enjoying an extended fifth cycle as a sedative agent. As an agent for general anesthesia, nitrous oxide is slowly losing popularity, however, for reasons described subsequently and in Chapter 18.
COMMON TERMS
GOALS OF ANESTHESIA
General anesthesia may be defined as “a drug-induced reversible depression of the CNS resulting in the loss of response to and perception of all external stimuli.”4 In practice, this simple definition is inadequate because it neglects the contributions of unconsciousness, amnesia, immobility, and autonomic stability to the anesthetic state and the fact that general anesthetics differ significantly in the effects they achieve.
The goals of anesthesia for general surgery also apply to dental surgery, but there are some important differences. Dental patients are generally outpatients; in most circumstances, particularly situations not involving extensive oral surgery, the procedures are not as traumatic as general surgical procedures, and it is neither necessary nor desirable to render the patient unconscious. Although general anesthesia is sometimes necessary, specific techniques have been developed for producing sedation in dental patients (see Chapter 48).
MECHANISMS OF ANESTHESIA
Molecular Mechanisms of Action
Correlates of anesthetic potency
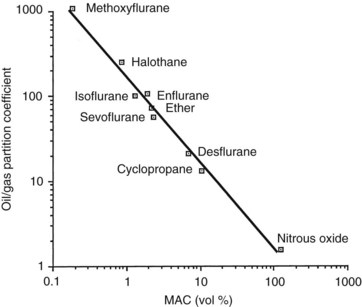
Various mechanistic theories of general anesthesia began to appear shortly after the landmark demonstration of ether-induced insensibility by Morton, but the first important observation was made independently by Meyer in 1899 and Overton in 1901, who emphasized the correspondence between the lipid solubility of an agent and its anesthetic potency (Figure 17-1). The Meyer-Overton correlation suggested that anesthesia begins when any chemical substance has attained a certain molar concentration in the hydrophobic phase of the cell membrane. When olive oil is used to represent the hydrophobic medium, this concentration is approximately 50 mmol/L. Experiments with different lipid media indicate that the best fit between solubility and anesthetic potency is obtained with lipids that are amphophilic (i.e., they have polar and nonpolar attributes) and can serve as hydrogen bond acceptors. These characteristics are descriptive of membrane phospholipids and cholesterol.
Membrane lipid theories
Numerous investigators since Mullins have sought to link the notion of a critical number or volume of anesthetic molecules with plasma membrane disturbances that could result in general anesthesia. Until the early 1980s, most attention was directed at the lipid bilayer of the plasma membrane, specifically the ability of anesthetics to cause membrane expansion, lipid fluidization, or lateral phase separation. With each of these effects, it was postulated that, as a result of the alteration in the lipid bilayer, the neuronal membrane becomes unable to facilitate the changes in protein configuration that are required for such essential steps in the transmission of nerve impulses as ion gating, synaptic transmitter release, and binding of the transmitter to the receptor.31
The membrane expansion theory was a natural outgrowth of the critical volume hypothesis. It holds that the absorption of anesthetic molecules by the lipid phase causes the membrane to expand, preventing important intrinsic membrane constituents from functioning properly. Measurements indicate that the expansion associated with general anesthesia is approximately 0.4%. Fluidization, or disordering, of lipids by anesthetic agents was noted in studies of lipid bilayers prepared with phospholipid and cholesterol to mimic cell membranes. Parallel shifts in measures of lipid fluidization and the activity of membrane-bound enzymes suggested that this perturbation of the normal lipid structure may result in functional changes sufficient to disrupt nerve transmission. The lateral phase separation theory was based on the idea that membrane lipids exist in two states: a high-volume, disordered sol state and a compact, ordered gel state.31 The ability of lipids to convert from the sol to the gel configuration, or to be compressed laterally within the membrane, was thought to accommodate conformational changes that need to occur for the opening of ion channels.
These lipid perturbation theories were supported by findings that hyperbaric pressures and certain convulsant drugs antagonize anesthesia, presumably by reversing membrane expansion or re-establishing order. It is now understood, however, that pressure or drug reversal of anesthesia arises from a physiologic antagonism of anesthetic action brought on by independent neurologic stimulation. Different anesthetics are affected differently by the same pressure, including chloral hydrate, whose anesthetic effect is immune to pressure reversal. Evidence has also mounted to cast doubt on membrane expansion or lipid perturbation per se as a cause of anesthesia. Direct measurements of the expansion of lipid bilayers and red blood cell membranes in response to anesthetic concentrations of ethanol and halothane yield values that are effectively insignificant, and other measurements have shown nonanesthetic long-chain alcohols to cause membrane expansion similar to that of inhalation anesthetics. Regarding fluidization or sol-gel transformations, changes equivalent to those associated with anesthesia can be attained by temperature elevations less than 1° C.32
Mechanisms involving membrane proteins
Membrane proteins constitute a second hydrophobic environment with which anesthetic molecules may interact.9 The idea that membrane proteins are the targets of anesthetic action is attractive for several reasons. First, it is consistent with the mode of action of most drugs that influence the CNS. Second, allosteric selection (described in Chapter 1) of a protein conformation by the binding of even a single small molecule can have pronounced effects on protein function. Third, it can best explain differences in action among the various anesthetics by assuming that these agents exert different effects on the same protein or influence different proteins altogether.
Although technical difficulties have long inhibited direct examination of anesthetic drug interactions with membrane proteins, the firefly enzyme luciferase provided a good initial model for study.7 Luciferase is a water-soluble protein that produces light when it cleaves its substrate, luciferin. For a wide variety of agents, anesthetic potency correlates directly with the ability to inhibit luciferin binding and prevent light emission. The binding site on the enzyme is amphophilic in nature and capable of accepting a hydrogen bond. Similarly, numerous potential sites exist for direct anesthetic interactions with proteins: hydrophobic regions within globular or folded polypeptides, between polypeptides joined in an oligomeric structure, and at the protein-lipid or protein-water interface.
The synthesis of a water-soluble four-stranded α-helical protein bundle similar in structure to peptides forming ligand-gated ion channels has allowed characterization of direct anesthetic-protein interactions.14 A hydrophobic cavity within the bundle binds halothane best when the cavity is lined with methionine and aromatic acid residues. Halothane binding stabilizes the protein in a conformation that putatively promotes anesthesia.
A close correspondence between the anesthetic potencies of stereoisomers of halothane and isoflurane and their ability to perturb ion channel function provides strong evidence that these membrane proteins are the immediate targets for general anesthetic action. It is now firmly established that certain classes of general anesthetics inhibit or activate specific ligand-gated ion channels in clinically relevant concentrations. Binding studies indicate a specific active site for volatile anesthetics on neuronal nicotinic receptors.2,19 Specific mutations on the M2 domains of the nicotinic receptor, which correspond to the α-helix segments that form the aqueous pore of the receptor’s ion channel, enhance the blocking action of anesthetics such as isoflurane and alcohols such as octanol.5 It is postulated that hydrophobic general anesthetic agents bind at a discrete site in the vicinity of the mutation loci. The more polar alcohols gain access to the same site preferentially after channel opening, suggesting that the binding site is within the channel itself. Inhibition of nicotinic receptors in skeletal muscle probably contributes to the ability of volatile anesthetics to enhance muscle relaxation. Actions at neuronal nicotinic receptors promote effects such as amnesia, hyperalgesia, and excitation observed at subanesthetic concentrations of volatile anesthetics and barbiturates.
The γ-aminobutyric acidA (GABAA) receptor has been implicated in the CNS depressant effect of most anesthetic drugs.4,30 Specific binding sites for benzodiazepines, barbiturates, other intravenous anesthetics, and volatile anesthetics have been described.4 Stimulation of these receptor sites increases the activity of GABA at its own separate site; many agents other than benzodiazepines can also open the GABAA Cl− channel in the absence of GABA. Hyperpolarization of the affected neuron inhibits neuronal activity. Glycine receptors constitute another group of inhibitory receptors that are activated by at least some general anesthetics (inhalation anesthetics, alcohols, thiopental, and propofol) in clinically relevant concentrations.
Excitatory receptors blocked by specific anesthetic agents include N-methyl-d-aspartate (NMDA), kainate, and α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptors. Ketamine and nitrous oxide13 selectively inhibit NMDA receptors, whereas barbiturates and certain inhalation anesthetics block AMPA and kainate receptors.4
In addition to the classic ligand-gated ion channels described previously, other ion channels may be involved in the actions of specific general anesthetics. Several types of 2-pore-domain K+ channels (identified by their acronyms TREK1, TREK2, TASK1, TASK3, and TRESK) are variably activated by inhalation anesthetics.4,6 These channels are responsive to intracellular second messengers and are believed to regulate background neuronal excitability and neurotransmitter release. Several types of Ca++ channels and Na+ are inhibited by clinical concentrations of drugs and may contribute an inhibitory influence on neurotransmitter release.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


