Congenital Genetic Disorders and Syndromes
 Outline
OutlineBasic Genetic Concepts
A person’s genome is made up of the DNA in all 46 chromosomes in the nucleus of each cell of the body. Each cell has 23 pairs of chromosomes. One chromosome of each pair is inherited from each parent. Two of the chromosomes are called sex chromosomes (X and Y), whereas the remaining chromosomes (numbered 1 through 22) are called autosomes. Males have one X and one Y chromosome; females have two X chromosomes. In each cell of a female, one of the X chromosomes is randomly inactivated. This is an important determinant of the severity of X-linked genetic disorders, as will be discussed later. The tool used to look at all of the chromosomes in a cell and to determine the sex of a fetus from amniotic fluid is called a karyotype (Figure 16-1). This technique also identifies major chromosomal anomalies such as trisomy (an extra chromosome), translocations of one part of a chromosome to another, or large chromosomal deletions.
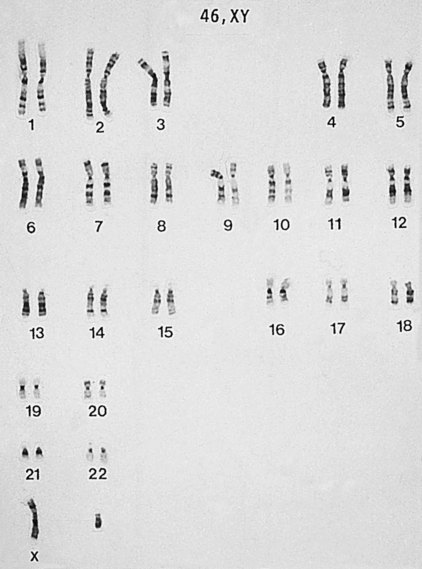
 FIGURE 16-1 Karyotype of a normal male.
FIGURE 16-1 Karyotype of a normal male.
Inheritance Patterns
Autosomal Recessive
An autosomal recessive disorder is only manifest when an individual has two copies of the mutant gene. Most frequently, each parent has one copy of the defective gene and is a carrier, and there is a 25% chance that both mutant genes will be passed on to their offspring. It is equally likely that males and females will be affected. Fifty percent of the time the offspring will get one copy of the mutant gene from one parent and will be a carrier, and 25% of the time the offspring will get two normal copies of the gene. Although autosomal recessive disorders are relatively uncommon, the carrier status in certain populations can be significant. For example, 1 in 25 people of northern European descent are carriers of cystic fibrosis.1
Multifactorial Inheritance
Most common diseases of adulthood (such as diabetes, hypertension, and manic depression) as well as most congenital malformations (cleft lip/palate and neural tube defects) are the result of multiple genes and gene-environment interactions, rather than a single gene defect. This is also true for the most common dental diseases (periodontal disease and dental caries). Multifactorial traits are thought to result from the interaction between multiple genes with multiple environmental factors. The most convincing evidence for this type of inheritance comes from twin studies. If a trait is multifactorial with a significant genetic component, monozygotic (identical) twins will both have the disease significantly more frequently than dizygotic (fraternal) twins. This has been demonstrated in multiple studies for dental caries among twins raised apart and provides strong evidence that there is a genetic component to dental caries susceptibility.2,3 More recently, researchers have completed a genome-wide association study to identify genetic loci associated with the susceptibility or resistance to dental caries.4
Dentist as Dysmorphologist
There are four basic mechanisms that result in structural defects during development. The first is malformation, the second is deformation due to mechanical forces, the third is disruption where there is a breakdown of tissues that were previously normal, and the fourth is dysplasia. Dysplasia is caused by a failure of normal organization of cells into tissues. It is not uncommon for humans to have at least one “minor” malformation. This includes things such as hair whorls, inner epicanthal folds, aberrant positioning of oral frenula, or preauricular pits. Although the occurrence of single anomalies such as these are relatively common and often present as a familial trait, there are a number of studies that have demonstrated that a child who has three minor anomalies has a much greater chance of having a major anomaly such as a defect in brain or heart development.5–7 This illustrates why it is important for health care professionals to be careful observers of their patients and to be familiar with the facial features that are considered to be normal or aberrant.
Dentists are trained to observe and examine the mouth, face, and other craniofacial structures. Coincidentally, many inherited diseases in humans involve malformations of the craniofacial region. Accurate diagnosis of developmental anomalies and their related disorders relies on the ability of the clinician to recognize and differentiate between normal and dysmorphic physical characteristics. According to the text Smith’s Recognizable Patterns of Human Malformation, 12 of the 26 categories of malformations used for diagnostic purposes involve features of the head or neck.8 Several are limited to oral structures, such as hypodontia, microdontia, micrognathia, and cleft lip/palate. In addition, having an understanding of the full spectrum of malformations associated with certain syndromes is essential for the safe and effective treatment of patients with these disorders.
Ocular Anomalies
Minor anomalies that affect the eyes and ocular region include widely spaced eyes (hypertelorism) (Figure 16-2), inner epicanthal folds (Figure 16-3), slanting of the palpebral fissures (upward or downward) (Figures 16-4 and 16-5), a single eyebrow (synophrys), blue sclera, and coloboma of the iris (cat-eye) (Figure 16-6).
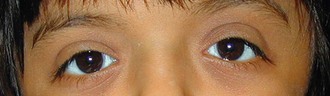
 FIGURE 16-2 Hypertelorism.
FIGURE 16-2 Hypertelorism.

 FIGURE 16-3 Inner epicanthal fold.
FIGURE 16-3 Inner epicanthal fold.
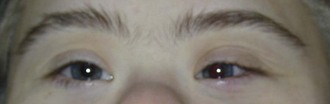
 FIGURE 16-4 Up-slanting palpebral fissures.
FIGURE 16-4 Up-slanting palpebral fissures.
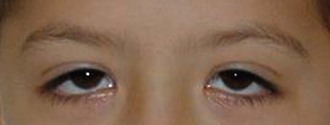
 FIGURE 16-5 Down-slanting palpebral fissures.
FIGURE 16-5 Down-slanting palpebral fissures.
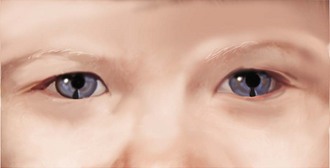
 FIGURE 16-6
FIGURE 16-6Auricular Anomalies
There are a number of minor anomalies that affect the outer ear (auricle) and the preauricular region. These include preauricular tags or pits (Figure 16-7), low-set and malformed ears (Figure 16-8), protruding ears, and slanted ears.
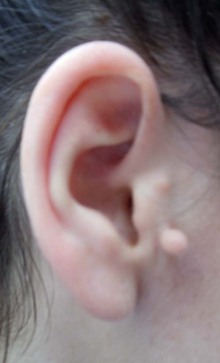
 FIGURE 16-7 Ear tag.
FIGURE 16-7 Ear tag.
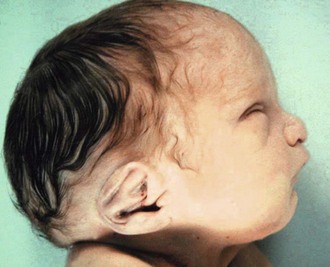
 FIGURE 16-8
FIGURE 16-8Anomalies of the Mouth and Oral Region
Cleft lip alone or combined with a cleft palate, although not a minor anomaly, can occur independently from other malformations and is then considered nonsyndromic. Other anomalies in this region include lower lip pits (Figure 16-9), bifid uvula, macroglossia, and prominent or full lips. Attached frenula as is seen with ankyloglossia (Figure 16-10) is also a fairly common anomaly/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


