CHAPTER 15 Anti-Parkinson Drugs*
Prodromal symptoms such as orthostatic hypotension, urinary retention, constipation, incontinence, excessive sweating, and pain are harbingers of possible Parkinson’s disease development. Loss of olfaction for particular odors (e.g., gasoline, banana, pineapple, smoke, cinnamon)19 appears before any motor abnormality develops. Disorders of the autonomic nervous system are common. Subtle changes in motor function may occur, such as changes in speech, handwriting changes, or reduced arm swinging during walking. The patient may have various ill-defined sensory symptoms consisting of numbness, tingling, abnormal temperature sensation, and visual disturbances.8 Higher rates of anxiety disorder (40%) are reported in Parkinson patients (e.g., panic disorder, generalized anxiety, and social phobia), and the anxiety issues may precede the motor symptoms.57 Pain and chronic pain are common in Parkinson’s disease, primarily related to dyskinesias66 but may also represent diseased central sensorimotor interactions.59 Sleep disturbances, including increased daytime sleepiness, abnormal rapid eye movement (REM) sleep, and increased motor activity during REM sleep (e.g., restless legs syndrome [RLS]), have been identified. Drooling commonly occurs and is mainly caused by difficulty in swallowing (dysphagia) rather than excessive salivation. Dysphagia may be severe and can result in death by asphyxia, aspiration, or pneumonia.
Although Parkinson indicated that the senses and intellect were “uninjured” in parkinsonism, the disease is associated with mental slowing that can be reversed with treatment. In addition, the incidence of dementia is approximately three times greater in patients with Parkinson’s disease than in age-matched control subjects. Symptoms of depression may develop.8 Approximately 30% of patients report hallucinations or delusions.
Parkinson’s disease may be primary, secondary, or Parkinson’s plus. The primary or idiopathic form is the typical elderly-onset disorder with motor signs and primary damage to the dopamine cells in the basal ganglia. Idiopathic Parkinson’s disease has been associated in patients older than 50 years, and the incidence increases with increasing age. The degenerative progression of idiopathic Parkinson’s disease proceeds at irregular rates, and a subclinical form of the disorder may exist for years before the patient has the motor signs associated with Parkinson’s disease. Higher incidences are seen in certain occupations, such as physicians, dentists, teachers, technicians, and agricultural workers.34 Some cases of primary Parkinson’s disease may have a genetic contribution and are usually seen in patients younger than 50 years. Candidate loci have been identified on chromosomes 1, 2, 4, 6, and 12.7 Secondary Parkinson’s disease is preceded by cerebral infections (syphilis, influenza), toxic chemicals (e.g., carbon monoxide, manganese, pesticides), cerebral hypoxia (vascular defects), traumatic brain injury,33 or antipsychotic drugs. The best-documented infectious source was the influenza epidemic of the late 1910s, which contributed to a delayed epidemic of cases as the patients aged.
Parkinson’s plus disorders are a group of maladies in which the signs and symptoms of Parkinson’s disease contribute to a larger disorder (e.g., multiple system atrophy) that often involves more extensive brain damage in the brainstem and cerebral cortex. In many cases, the exact cause of the parkinsonism is unknown. Certain activities and chemicals may reduce the risk of Parkinson’s disease, including physical exercise, statin anticholesterol drugs, low-fat diets, smoking,22,64 caffeine (coffee, tea),38 and moderate alcohol consumption.
NEUROBIOLOGY AND PATHOPHYSIOLOGY
Lewy bodies are neuron inclusion bodies found in the brains of patients with idiopathic Parkinson’s disease and some other neurologic disorders. These inclusion bodies contain α-synuclein, ubiquitin, and in some cyclin-dependent kinase 5 (cdk5) and leucine-rich repeat kinase 2 (LRRK2).7 The role of α-synuclein has been associated with synapses and nuclei (“sy” + “nuclei” + “n”), cell membrane, mitochondrial membranes, and possibly reuptake transporters.2 Excess formation of α-synuclein is termed a synucleinopathy. Ubiquitin is associated with the ubiquitin-proteasome catabolic system involved with removal of damaged or misfolded proteins (e.g., synuclein). The cdk5 enzyme has a role in regulating microtubular structural proteins that mediate neuronal function. The accumulation of cdk5 also may contribute to inflammatory changes in the brain.17 LRRK2 has been found in some genetic Parkinson’s disease cases in Asia. Lewy bodies decrease neuron survival of the affected cells, which may trigger increased microglial mediated inflammation and cell destructive activity.52
By studying the pattern of α-synuclein inclusions, it has been observed that the common sporadic form of Parkinson’s disease starts in the brainstem and olfactory tract before motor symptoms appear. The most sensitive neurons apparently are small neurons with long axons, typical of catecholamine and other autonomic neurons. Early damage occurs in the dorsal motor nucleus of the vagus, spinal cord,11 and olfactory neurons. Prodromal autonomic and olfactory symptoms coincide with the development and spread of α-synuclein accumulations. As the disorder progresses, the lesions ascend to involve brainstem, thalamus, basal ganglia, limbic, and finally cortical structures.10 When sufficient damage to dopamine cells occurs, clinical motor signs become evident. The overt symptoms generally progress over 10 to 20 years and may terminate in severe invalidism. Life expectancy is reduced because of the usual complications associated with long-term invalidism. There is growing interest in developing neuroprotective treatments that might be useful before these debilitating changes occur.
Hereditary Parkinson’s diseases are familial disorders usually seen in patients younger than 50 years old. Most of the known genetic differences that predispose to Parkinson’s disease are associated with changes in α-synuclein, ubiquitin-proteosome, or mitochondrial function; the number of possibilities continues to increase with further research (Table 15-1). Autosomal dominant loci tend to induce the disease, whereas recessive factors often contribute to expression but may not act alone. Mutations that affect the function of dopamine neurons can cause Parkinson’s symptoms directly, whereas in other cases Lewy bodies are conspicuous and may lead to subsequent damage of the dopamine cells.

Environmentally induced Parkinson’s disease is a result of a toxic agent from the environment or brain trauma that damages the basal ganglia. A dramatic upsurge of interest in environmental factors as a cause for Parkinson’s disease occurred when several young drug abusers developed severe parkinsonian symptoms after self-administering a drug that they thought was a heroin analogue. The compound, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), was a by-product of a faulty synthesis of the opiate analgesic alphaprodine. Later work established that the chemical causing the toxicity was actually 1-methyl-4-phenylpyridinium (MPP+), a metabolite of MPTP. MPP+ toxicity stems from its transportation into cells by catecholamine reuptake transporters and subsequent interference with mitochondrial energy production. Acute MPP+ selectively destroys dopamine neurons in the substantia nigra, and this replicates many of the motor disorders of Parkinson’s disease, although not the synucleinopathy.4 In some models, chronic or continuous MPTP administration can compromise the ubiquitin-proteasome system and can lead to the accumulation of α-synuclein.24 MPTP-induced damage is reduced in α-synuclein knockout mice, suggesting that these inclusions may potentiate the toxin’s damage.40
Various environmental agents may contribute to the development of Parkinson’s disease, and various pyridines are present in the environment in insecticides, herbicides, and contaminants. The insecticides rotenone and paraquat (structurally related to MPP+) have been found to produce a Parkinson-like syndrome in rats.51 Antipsychotic drugs often produce reversible Parkinson-like signs. The antipsychotic agent haloperidol is metabolized to a pyridinium metabolite that increases oxidative damage in experimental animals,39 raising the possibility of irreversible damage. Mitochondrial respiratory complexes may be compromised by MPP+, and mitochondrial complex 1 has been found to be prone to functional compromise. It is unclear whether mitochondrial disorders are a feature of idiopathic Parkinson’s disease, however.36 Another important environmental toxin is the metal manganese, and miners and users of some manganese products may develop secondary Parkinson’s disease.
Changes in Brain Function in Parkinson’s Disease
Motor function in the spinal cord and trigeminal nucleus is directly controlled by motor regions of the cerebral cortex. A model of basal ganglia function is shown in Figure 15-1. Motor activity in the cerebral cortex is stimulated by thalamocortical input, which is modulated by many factors, including sensory feedback, memory readout, and emotional response. The basal ganglia modulate the thalamocortical processing through a γ-aminobutyric acid (GABA)–mediated inhibitory input derived from the substantia nigra pars reticulata and the internal part of the globus pallidus, nuclei whose functions seem to be similar and for this discussion are referred to as the basal ganglia thalamic inhibitor (BGTI). The output of the BGTI is influenced by two pathways within the basal ganglia, the direct and indirect. The direct pathway generally inhibits the BGTI and disinhibits the thalamic drive and increases motor activity. The indirect pathway, involving excitatory input from the subthalamic nucleus, inhibits movement by facilitating BGTI outflow.
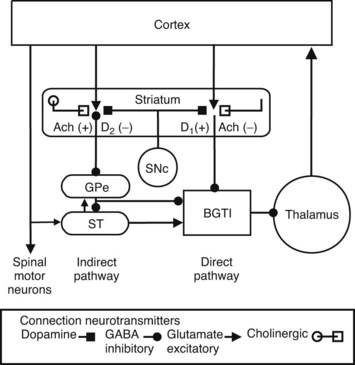
Dopamine from the substantia nigra pars compacta (SNc) innervates the direct and indirect pathways in the striatum.4 In the direct pathway, it acts on D1 receptors, which increase GABA inhibition of the BGTI. In the indirect pathway, dopamine acts on D2 receptors, which inhibit GABA outflow to the indirect pathway. In the case of Parkinson’s disease, a loss of dopamine neurons from the SNc leads to dysregulation of the indirect pathway by the subthalamic nucleus, producing activation of the BGTI and inhibition of movement (bradykinesia or akinesia). Damage anywhere in these loops may result in localized motor abnormalities such as dystonias. Parallel cortico–basal ganglia–thalamic circuits are proposed that subserve aspects of executive and emotional thinking, which may help explain some of the psychiatric symptoms (e.g., depression, hallucinations) observed in some patients with Parkinson’s disease.47,54
The tremor associated with Parkinson’s disease has been more difficult to explain. Research has suggested that the interaction between the subthalamic nucleus and the external segment of the globus pallidus may be one source of tremor. Other studies suggest a role for the thalamus in tremor generation. Tremors may also represent a dysregulation of the communication between the cerebellum and basal ganglia and other parts of the brain. Other research has found a relationship between acetylcholine, norepinephrine, or serotonin18,25 levels and tremors.
Parkinson’s tremors predominate when the patient is resting, whereas cerebellar loop damage tremors predominate during movements. Cortical-striatal-thalamic loops, impaired in Parkinson’s disease, are associated with deficits in internally guided motor activity—difficulty with volitional behavior. A second motor control circuit, the cerebellar-thalamic-cortical loop, is thought to be more important for reacting to externally guided behavior. Evidence has accumulated more recently that these systems communicate at many levels—cortical,46 thalamic,37 and midbrain.3
Hints for a role of the cerebellum in Parkinson’s disease include the observation that patients can sometimes initiate blocked motor behaviors when presented with externally applied cues, such as an external target to guide a foot to initiate walking. In addition, patients with Parkinson’s disease freeze in conjunction with certain external stimuli, such as passing through a doorway. Functional brain imaging suggests that the cerebellum is hyperactive in patients with Parkinson’s disease and possibly trying to compensate for the basal ganglia deficiency.20,42
Although Parkinson’s disease is primarily associated with movement problems, there may be additional changes. Responses to sensory stimuli, measured in the basal ganglia, tend to have large receptive fields. This tendency suggests that the basal ganglia may participate in poorly localized sensations such as pain.60 As noted earlier, changes in olfaction and pain are early signs of the disorder. Pain sensation may be altered in Parkinson’s disease, including reductions in discriminative, affective, and cognitive dimensions.14 Patients differ on the direction of the sensory changes. In some cases, pain responding may be reduced, whereas in others diffuse and annoying spontaneous pains may be present, often associated with muscle pain. The basal ganglia may participate in the actions of opiates because the globus pallidus is rich in opiate receptors.6
Neuroprotection
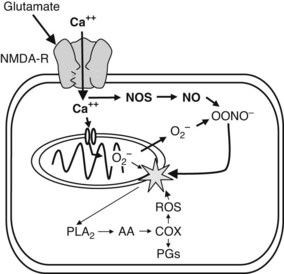
Neuroprotective strategies for the treatment of Parkinson’s disease have evolved from theories regarding the cause of the disease. The free radical hypothesis is based on the concept that free radicals (generated from oxidative reactions) react with membrane lipids and cause lipid peroxidation, cell injury, and subsequent cell death. Mitochondrial damage and inhibition of oxidative phosphorylation also occur as a result of free radical attack.9 Figure 15-2 illustrates the relationship between dopamine metabolism and the generation of reactive oxygen species. In addition, the damaged neurons may lose resistance to the toxic effects of the afferent excitatory neurotransmitter glutamate (afferents from the cortex, pedunculopontine nucleus, and subthalamic nucleus), whose toxicity is mediated by excessive Ca++ flux and nitric oxide synthesis (Figure 15-3). In the most severe case, these reactions can lead to apoptosis and cell death.
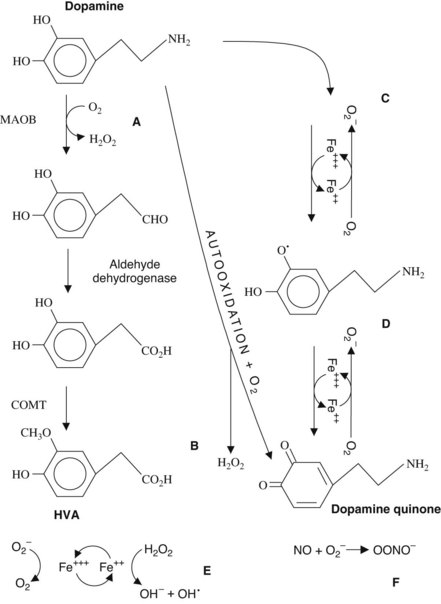
Free radical scavenger drugs such as vitamin E have been administered experimentally, albeit so far without much success. The monoamine oxidase (MAO) B inhibitor selegiline offers a potential, but as yet unproved, neuroprotective action by inhibiting the breakdown of dopamine. The pituitary hormone melatonin has antioxidant and proglutathione actions and has been found to be protective in the MPP+ model. Other vitamins may influence neuroprotective processes. Vitamin A and D receptors (RXR and VDR) are concentrated in the substantia nigra, and deficiencies may contribute to Parkinson symptoms. The NURR1 genetic defect affects the RXR receptor. Inadequate levels of vitamins A and D may potentiate the degree of Parkinson symptoms in addition to increasing the risks of bone fractures associated with falls.49,63
Late-stage Parkinson’s disease is quite resistant to treatment; drugs become ineffective, and tissue grafts and implants lose efficacy. Examples of irreversible neural degenerative cascades45 may provide a framework for understanding why Parkinson’s disease continues to progress (pathologic remodeling, gliosis, or vascular problems) despite attempts to replace dopamine. Earlier interventions that could prevent such irreversible changes are needed.
Tomographic Imaging in Parkinson’s Patients
Tomographic imaging techniques are useful for investigations into causes, disturbed brain processing, and diagnostic information about Parkinson’s disease.41 Systems used to image brain changes in patients with Parkinson’s disease include positron emission tomography; single photon emission computed tomography, which can assess ligand binding within the brain; and functional magnetic resonance imaging (fMRI) and the related blood oxygen level–dependent (BOLD) fMRI. These all are indirect measures of brain tissue function.16 Transcranial ultrasound has been useful for finding changes in substantia nigra iron levels. Table 15-2 summarizes several imaging ligands and how they are used to assess the function of the basal ganglia and other related brain regions. In particular, β-CIT has been used to visualize dopamine uptake transporters in the basal ganglia. These images distinctly outline the caudate-putamen region of the brain and show that in Parkinson’s disease the labeling in the basal ganglia is reduced either unilaterally or bilaterally.
TABLE 15-2 Imaging Techniques Used to Evaluate Parkinson’s Disease
| NAME | USES |
|---|---|
| 18Fluoro-DOPA | Labels levodopa-to-dopamine conversion and presynaptic dopamine |
| Beta-CIT, (2 beta-carbomethoxy-3 beta-(4-iodophenyl)tropane) | Selectively labels DAT |
| SCH 39166 and NNC 756 | Selective D1 receptor binding |
| Raclopride | Selectively binds to dopamine D2 receptors |
| d-Methylphenidate | Sensitive to dopamine system loss in basal ganglia |
| WAY100635 | Labeling 5-HT1A, shown to be inversely related to Parkinson’s tremor |
| Fluorodeoxyglucose | Labels glucose (energy) use for differential diagnosis between Parkinson’s disease and MSA |
| 15Oxygen | PET oxygen use technique for assessment of Parkinson’s disease and MSA |
| PK11195 | PET marker of PBBS, which are selectively expressed by activated microglia associated with brain inflammation |
| fMRI and BOLD fMRI | Indirect activity measure based on blood desaturation in brain active regions. Used to assess relative activity in different brain regions. May be useful to assess MSA |
| Transcranial ultrasound | Used to identify iron accumulation in substantia nigra in pre–Parkinson’s disease or early Parkinson’s disease |
Positron emitters include 123I, 18F, or 11C.
5-HT1A, 5-Hydroxytryptamine type 1A; BOLD, blood oxygen leveldependent; DAT, dopamine transporter; fMRI, functional magnetic resonance imaging; MSA, multiple system atrophy; PBBS, peripheral benzodiazepine sites; PET, positron emission tomography.
Unified Parkinson’s Disease Rating Scale
Using only Part III, Motor Examination, an individual without Parkinson’s disease would score 0 or a low value. For a Parkinson’s patient whose symptoms have been reduced with treatments and who feels “on” (i.e., able to move freely), the score is less than 8. Patients feel “off” when the score is greater than 20.13 One sample of Parkinson’s patients65 averaged around 41 in the untreated state and around 22 in the group with levodopa treatment. Some of these patients also received subthalamic nucleus deep brain stimulation, and their scores also decreased about 20 points on deep brain stimulation alone. There is considerable variation in responding, such that one patient may improve from 56 to less than 8, whereas another patient may show no improvement or worsening. Some patients respond to placebo with improvement of motor function.32 Over time without treatment, a typical patient’s score increases about 6 points per year as the disease progresses.23 Published UPDRS scores for several Parkinson’s disease treatments are summarized in Table 15-3.
TABLE 15-3 Selected Anti-Parkinson Drugs and Their Approximate Effect on Unified Parkinson’s Disease Rating Scale (UPDRS) Motor Examination Scores
| AGENT | REDUCTION OF UPDRS III |
|---|---|
| Levodopa | 22 |
| Deep brain stimulation | 20 |
| Apomorphine* | 10 |
| Bromocriptine | 5 |
| Pergolide | 5 |
| Pramipexole | 4 |
| Ropinirole | 5 |
| Rotigotine | 5 |
| Amantadine | 6 |
| Selegiline | 2 |
| Rasagiline | 5 |
| Tocopherol (vitamin E) | 0 |
| Melatonin | 0 |
| Placebo | 0-5 |
DOPAMINE REPLACEMENT AS A BASIS FOR THERAPY
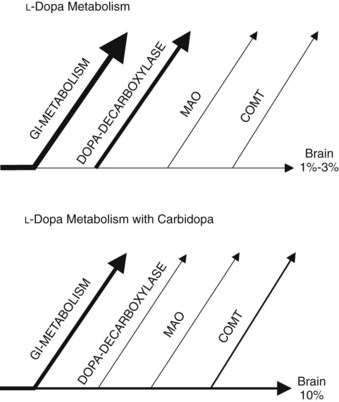
Carbidopa, a dopa-decarboxylase inhibitor, prevents much of the peripheral metabolism of levodopa (Figure 15-4). Carbidopa does not cross the blood-brain barrier, so it does not inhibit the CNS synthesis of dopamine from levodopa. Systemically administered levodopa is transported into the brain through the blood-brain barrier by a saturable amino acid transporter. Combining levodopa with carbidopa permits the use of lower doses of levodopa with greater effectiveness in parkinsonism compared with levodopa alone. It has been found that catechol-O-methyltransferase (COMT) may become upregulated in patients taking carbidopa. This discovery prompted the development of COMT inhibitors to inhibit further the peripheral metabolism of levodopa. Another enzyme important in catecholamine metabolism is MAO. MAO-B inhibitors may also help reduce the amount of levodopa required to reduce Parkinson’s symptoms.
Increased dopamine release in the basal ganglia is produced by such behaviors as eating food, listening to music,46 and sexual activity. In fear-mediating brain regions (e.g., amygdala), dopamine may act to make fearful stimuli more salient.2 Dopamine cells fire more during waking than sleep, suggesting a role in sleep-wake cycles. This relationship is complicated by the multiple dopamine receptors, some of which seem to promote sleepiness.48 Dopamine may modulate appetite, but again the relationship is not simple. Dopamine neurons inhibit the release of prolactin by an action on the hypothalamus. Dopamine is associated with the chemoreceptor trigger zone where it produces an emetic action. Dopamine neurons are also found in the eye and olfactory tract. Dopamine is also a precursor for the neurotransmitter norepinephrine and so may alter blood pressure or cardiovascular function. Dopamine has a vasodilatory action in the kidney. Similar to many neurotransmitters, dopamine has many and varied actions.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


