Impact of Periodontal Infection on Systemic Health
Knowledge of the pathogenesis of periodontal diseases has evolved markedly over the last 50 years.65 Periodontal disease is an inflammatory disease initiated by bacterial pathogens. Environmental, physical, social, and host stresses may affect and modify disease expression through a multitude of pathways. Certain systemic conditions can affect the initiation and progression of gingivitis and periodontitis (see Chapters 6 and 11). Systemic disorders that affect neutrophil, monocyte, macrophage, and lymphocyte function result in the altered production or activity of host inflammatory mediators.65,102 These alterations may manifest clinically as the early onset of periodontal destruction or as a more rapid rate of destruction than would occur in the absence of such disorders.
Pathobiology of Periodontitis
Our understanding of the pathogenesis of periodontitis has changed remarkably over the last 30 years.65,102,106 The nonspecific accumulation of bacterial plaque was once thought to be the cause of periodontal destruction, but it is now recognized that periodontitis is an infectious disease associated with a small number of predominantly gram-negative microorganisms that exist in a subgingival biofilm.40 Furthermore, the importance of the host in disease initiation and progression is clearly recognized. Although pathogenic bacteria are necessary for periodontal disease, they are not sufficient alone to cause the disease. A susceptible host is also imperative. In a host who has relatively low susceptibility to disease, bacterial pathogens may have no clinical effect. This may be due to a particularly effective host immunoinflammatory response that eliminates pathogenic organisms while minimizing destruction of native tissues. Conversely, in a host with relatively high disease susceptibility, the marked destruction of periodontal tissues may result.
There are many systemic conditions that can modify the host’s susceptibility to periodontitis. For example, patients with immune suppression may not be able to mount an effective host response to subgingival microorganisms, thereby resulting in more rapid and severe periodontal destruction. Conversely, individuals with a significant increase in the production of proinflammatory mediators may respond to periodontal pathogens with an exuberant inflammatory response that results in the destruction of periodontal tissues. Although the potential impact of many systemic disorders on the periodontium is well documented, recent evidence suggests that periodontal infection may significantly enhance the risk for certain systemic diseases or alter the natural course of systemic conditions.89,120 Conditions in which the influences of periodontal infection are documented include coronary heart disease (CHD) and CHD-related events such as angina, infarction, atherosclerosis, and other vascular conditions; stroke; diabetes mellitus; preterm labor, low-birth-weight delivery, and preeclampsia; and respiratory conditions such as chronic obstructive pulmonary disease89,105 (Box 12-1).
Focal Infection Theory Revisited
Research in the area of periodontal medicine marks a resurgence in the concept of focal infection. In 1900, British physician William Hunter first developed the idea that oral microorganisms were responsible for a wide range of systemic conditions that were not easily recognized as being infectious in nature.97,135 He claimed that the restoration of carious teeth instead of extraction resulted in the trapping of infectious agents under restorations. In addition to caries, pulpal necrosis, and periapical abscesses, Hunter also identified gingivitis and periodontitis as foci of infection. He advocated the extraction of teeth with these conditions to eliminate the source of sepsis. Hunter thought that teeth were prone to septic infection primarily because of their structure and their relationship to alveolar bone. He stated that the degree of systemic effect produced by oral sepsis depended on the virulence of the oral infection and the individual’s degree of resistance. He also thought that oral organisms had specific actions on different tissues and that these organisms acted by producing toxins, thereby resulting in low-grade “subinfection” that produced systemic effects over prolonged periods. Finally, Hunter thought that the connection between oral sepsis and resulting systemic conditions could be shown via the removal of the causative sepsis through tooth extraction and observation of the improvement in systemic health. Because it explained a wide range of disorders for which there was no known explanation at the time, Hunter’s theory became widely accepted in Britain and eventually the United States, thereby leading to the wholesale extraction of teeth.
The focal infection theory fell into disrepute during the 1940s and 1950s, when widespread extraction—often of the entire dentition—failed to reduce or eliminate the systemic conditions to which the supposedly infected dentition had been linked.135 The theory, while offering a possible explanation for perplexing systemic disorders, had been based on very little (if any) scientific evidence. Hunter and other advocates of the theory were unable to explain how focal oral sepsis produced these systemic maladies. They were also unable to elucidate possible interactive mechanisms between oral and systemic health. Furthermore, the suggested intervention of tooth extraction often had no effect on the systemic conditions for which patients sought relief. However, Hunter’s ideas did encourage extensive research in the areas of microbiology and immunology.
Evidence-Based Clinical Practice
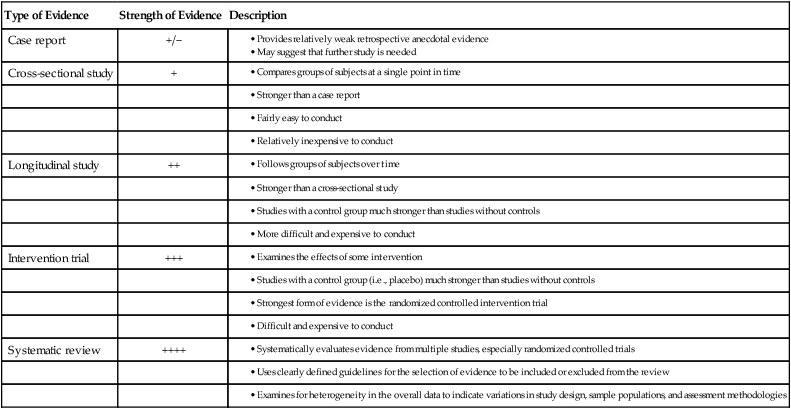
Many of the precepts of the focal infection theory are being revived today in light of recent research demonstrating links between oral and systemic health. However, for the “hypothesis not to fall into disrepute for a second time, there must be no unsubstantiated attributions, no theories without evidence.”97 Today’s era of evidence-based medicine and dentistry provides an excellent environment in which to examine the possible relationships between oral infection and systemic disorders. To establish a relationship between conditions A and B, different levels of evidence must be examined. All scientific evidence is not given the same weight.49,92,98 The stronger the evidence, the more likely it is that a true relationship exists between the conditions. Table 12-1 describes these various levels of evidence.
TABLE 12-1
| Type of Evidence | Strength of Evidence | Description |
| Case report | +/− |
Subgingival Environment as a Reservoir for Bacteria
The subgingival microbiota in patients with periodontitis provides a significant and persistent gram-negative bacterial challenge to the host that is met by a potent immunoinflammatory response.99 These organisms and their products, such as lipopolysaccharide (LPSs), have ready access to the periodontal tissues and to the circulation via the sulcular epithelium, which is frequently ulcerated and discontinuous. Even with treatment, the complete eradication of these organisms is difficult, and their reemergence is often rapid. The total surface area of pocket epithelium in contact with subgingival bacteria and their products in a patient with generalized moderate periodontitis has been estimated to be approximately the size of the palm of an adult hand, with even larger areas of exposure in cases of more advanced periodontal destruction.106 Bacteremias are common after mechanical periodontal therapy, and they also occur frequently during normal daily function and oral hygiene procedures.32,70,90 Just as the periodontal tissues mount an immunoinflammatory response to bacteria and their products, systemic challenge with these agents also induces a major vascular response.28,46,109 This host response may offer explanatory mechanisms for the interactions between periodontal infection and a variety of systemic disorders.
Periodontal Disease and Mortality
The ultimate medical outcome measure is mortality. A number of studies suggest that an increased mortality rate is associated with inflammatory periodontal diseases.30,34,55,68,113 The Normative Aging Study examined 2280 healthy men every 3 years for more than 30 years after baseline clinical, radiographic, laboratory, and electrocardiographic examinations. A subset of this population was examined in the Veterans Affairs Dental Longitudinal Study to determine age-related changes in the oral cavity and to identify risk factors for oral disease. Clinical examinations were performed, and alveolar bone level measurements were determined from full-mouth radiographs. The mean percentage of alveolar bone loss and the mean probing depth were determined for each subject.
A study of data from this subject population sought to determine whether periodontal disease status was a significant predictor of mortality independent of other baseline characteristics within the population.34 From the original sample of 804 dentate, medically healthy subjects, a total of 166 died during the study. Periodontal status at the baseline examination was a significant predictor of mortality independent of other factors, such as smoking, alcohol use, cholesterol levels, blood pressure, family history of heart disease, education level, and body mass. For those subjects with the most alveolar bone loss, who averaged more than 21% alveolar bone loss at baseline, the risk of dying during the follow-up period was 70% higher than for all other subjects. Interestingly, alveolar bone loss increased the risk of mortality more than smoking (52% increased risk), which is a well-known risk factor for mortality. A later evaluation of these same subjects confirmed a higher incidence of CHD-related events such as MI and unstable angina among men less than 60 years old with alveolar bone loss as compared with those without bone loss.30
In a prospective cohort study of 1400 dentate men from Northern Ireland, subjects were divided into thirds (tertiles) based on the average periodontal attachment loss.68 Those with the highest level of periodontal attachment loss has a significantly higher risk of death as compared with those with the least attachment loss. The mortality rate over a 9-year period was 15.7% in those with the greatest attachment loss as compared with 7.9% in subjects with the lowest level of periodontal attachment loss. In these studies, periodontitis preceded and increased the risk of mortality. However, this only establishes an association; it does not confirm causation. It is possible that periodontal disease reflects other health behaviors not evaluated in this study rather than acting as a specific cause of mortality. In other words, patients with poor periodontal health may also have other risk factors that increase mortality rate (e.g., smoking).
When examining research that suggests oral health status as a possible risk factor for systemic conditions, it is important to recognize when other known risk factors for those systemic conditions have been accounted for in the analysis. Host susceptibility factors that place individuals at risk for periodontitis may also place them at risk for systemic diseases such as cardiovascular disease. In these patients, the association may actually be among the risk factors rather than among the diseases. For example, periodontitis and cardiovascular disease share such risk factors as smoking, age, race, male gender, and stress. Genetic risk factors may also be shared.64 In the Veterans Affairs Dental Longitudinal Study, smoking was an independent risk factor for mortality. When examining the data to determine whether periodontal status was a risk factor, smoking status and other known risk factors for mortality were removed from the equation to allow for the independent evaluation of periodontal status. Other studies support the association between poor oral health and an increased risk for mortality.113 In a prospective longitudinal study of subjects with type 2 diabetes, those with severe periodontitis had 3.2 times the risk of death from ischemic heart disease or kidney disease as compared with subjects without periodontitis or with only slight periodontitis after adjusting for other risk factors, including age, sex, duration of diabetes, glycemic control, macroalbuminuria, body mass index, serum cholesterol concentration, hypertension, and current smoking.113
Periodontal Disease and Coronary Heart Disease/Atherosclerosis
To further explore the association between periodontal disease and CHD/atherosclerosis, investigators have studied specific systemic disorders and medical outcomes to determine their relationship to periodontal status. CHD-related events are a major cause of death. MI has been associated with acute systemic bacterial and viral infections, and MI is sometimes preceded by influenza-like symptoms.84,125 Is it possible that oral infection is similarly related to MI? Traditional risk factors such as smoking, dyslipidemia, hypertension, and diabetes mellitus do not explain the presence of coronary atherosclerosis in a large number of patients. Localized infection that results in a chronic inflammatory reaction has been suggested as a mechanism underlying CHD in these individuals.91
In cross-sectional studies of patients with acute MI or confirmed CHD who are compared with age- and gender-matched control patients, patients with MI had significantly worse dental health (e.g., periodontitis, periapical lesions, caries, pericoronitis) as compared with controls.54,80,83 This association between poor dental health and MI was independent of known risk factors for heart disease, such as age, cholesterol levels, hypertension, diabetes, and smoking. Because atherosclerosis is a major determinant of CHD-related events, dental health has also been related to coronary atheromatosis. Mattila and colleagues81 performed oral radiographic examinations and diagnostic coronary angiography on men with known CHD and found a significant correlation between the severity of dental disease and the degree of coronary atheromatosis. This relationship remained significant after accounting for other known risk factors for coronary artery disease. Similarly, Malthaner and colleagues79 found an increased risk of angiographically defined coronary artery disease in subjects with greater bone loss and attachment loss; however, after adjusting for other known cardiovascular risk factors, the relationship between periodontal status and coronary artery disease was no longer statistically significant. There is evidence that the extent of periodontal disease may be associated with CHD. For example, there may be a greater risk for CHD-related events, such as MI, when periodontitis affects a greater number of teeth in the mouth as compared with subjects who have periodontitis that involves fewer teeth.6
Cross-sectional studies thus suggest a possible link between oral health and CHD; however, such studies cannot determine causality in this relationship. Rather, dental diseases may be indicators of general health practices. For example, periodontal disease and CHD are both related to lifestyle and share numerous risk factors, including smoking, diabetes, and low socioeconomic status. Bacterial infections have significant effects on endothelial cells, blood coagulation, lipid metabolism, and monocytes and macrophages. The research of Mattila and colleagues80 showed that dental infections were the only factors—other than the classic and well-recognized coronary risk factors—that were associated independently with the severity of coronary atherosclerosis.
Longitudinal studies provide compelling data regarding this relationship. In a 7-year follow-up study of the patients from the study of Mattila and colleagues, dental disease was significantly related to the incidence of new fatal and nonfatal coronary events as well as to overall mortality.82 In a prospective study of a national sample of adults, subjects with periodontitis had a 25% increase in the risk for CHD as compared with those with no or minimal periodontal disease, after adjusting for other known risk factors.29 Among younger males between the ages of 25 and 49 years, periodontitis increased the risk of CHD by 70%. The level of oral hygiene was also associated with heart disease. Patients with poor oral hygiene, as indicated by higher debris and calculus scores, had a twofold increased risk for CHD.
In another large prospective study, 1147 men were followed for 18 years.10 During that time, 207 men (18%) developed CHD. When periodontal status at baseline was related to the presence or absence of CHD-related events during the follow-up period, a significant relationship was found. Subjects with more than 20% mean bone loss had a 50% increased risk of CHD as compared with those with up to 20% bone loss. The extent of sites with probing depths of more than 3 mm was strongly related to the incidence of CHD. Subjects with probing depths of more than 3 mm on at least half of their teeth had a twofold increased risk, whereas those with probing depths of more than 3 mm on all of their teeth had more than a threefold increased risk of CHD. This study and others in which the periodontal condition is known to have preceded the CHD-related events support the concept that periodontal disease is a risk factor for CHD, independent of other classic risk factors. Not all studies, however, support this concept; some show little independent effect of periodontal status on the risk for CHD after adjusting for commonly accepted cardiovascular risk factors.50,51 It is particularly difficult to control for smoking as a confounding variable in these studies, because it is such an important risk factor for both periodontal disease and cardiovascular disease. This confounding influence of smoking makes it difficult to clarify the significance of the relationship between the diseases.
Perhaps the best evidence available comes from systematic reviews of studies examining the relationship between periodontal infection and cardiovascular diseases. A systematic review and meta-analysis of data from 15 studies showed a significant 14% to 222% increase in the risk of CHD-related events in patients with periodontal disease as compared with those without.8 A similar systematic review of longitudinal cohort and case-control studies showed a significant increase in risk of incident MI, angina, or CHD-related death in subjects with periodontitis in five of the six studies reported.31 This increased risk was noted mainly in younger subjects (i.e., <65 years old). Janket and colleagues53 performed a meta-analysis of periodontal disease as a risk factor for future cardiovascular events and found an overall 19% increased risk of such events among individuals with periodontitis. The increase in risk was greater (44%) among people less than 65 years old. Although this increased risk is fairly modest, the extensive prevalence of periodontal disease in the population may increase the significance of the risk from a public health perspective. An extensive systematic review by Scannapieco and colleagues114 concluded that a moderate degree of evidence exists to support an association between periodontal disease and atherosclerosis, MI, and cardiovascular disease; however, causality is unclear. A comprehensive review of the evidence was recently performed by an American Heart Association working group.69 That group concluded that “periodontal disease is associated with atherosclerotic vascular disease independent of known confounders.” However, the working group cautioned that evidence of a causal link between periodontitis and vascular diseases did not yet exist. Importantly, insufficient evidence exists to show that the treatment of periodontal disease has any impact on the risk of heart disease. Intervention trials are needed to make this determination.
Effect of Periodontal Infection
Although a direct causal link between periodontal diseases and vascular diseases has not been shown to date, there are numerous mechanisms—both direct and indirect—through which periodontal infection may affect the onset or progression of atherosclerosis and CHD.59 Periodontitis and atherosclerosis both have complex etiologic factors that combine genetic and environmental influences. In addition to smoking, the diseases share many risk factors and have distinct similarities with regard to their basic pathogenic mechanisms.
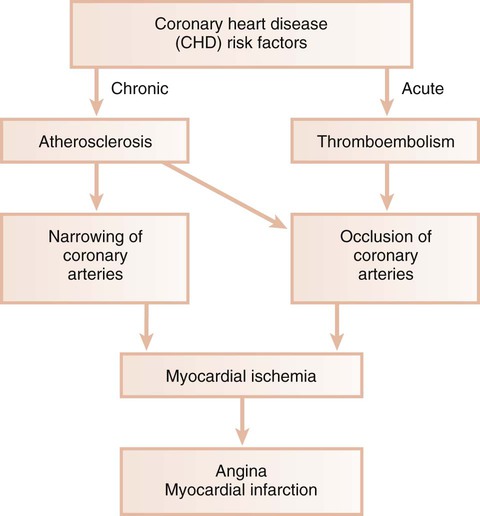
Ischemic Heart Disease.
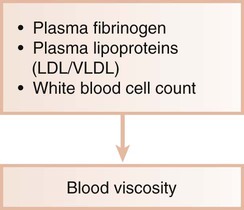
Ischemic heart disease is associated with the processes of atherogenesis and thrombogenesis (Figure 12-1). Increased viscosity of blood may promote major ischemic heart disease and cerebrovascular accident (stroke) by increasing the risk of thrombus formation.76 Fibrinogen is a major factor in the promotion of this hypercoagulable state. Fibrinogen is the precursor to fibrin, and increased fibrinogen levels increase blood viscosity. Increased plasma fibrinogen is a recognized risk factor for cardiovascular events and peripheral vascular disease77 (Figure 12-2). Elevated white blood cell count is also a predictor of heart disease and stroke, and circulating leukocytes may promote the occlusion of blood vessels. Coagulation factor VIII/von Willebrand factor has likewise been associated with the risk of ischemic heart disease.110
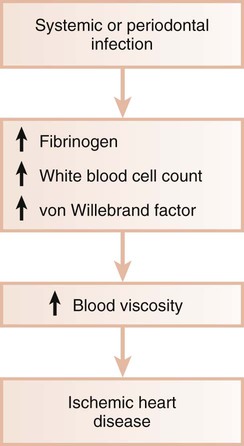
Systemic Infections.
Systemic infections are known to induce a hypercoagulable state and to increase blood viscosity (Figure 12-3). Fibrinogen levels and white blood cell counts are often increased in patients with periodontal disease.19,67 Individuals with poor oral health may also have significant elevations in coagulation factor VIII/von Willebrand factor antigen, thereby increasing the risk of thrombus formation. Thus, periodontal infection may also promote increased blood viscosity and thrombogenesis, which leads to an increased risk for central and peripheral vascular disease.
Daily Activity.
Routine daily activities such as mastication and oral hygiene procedures result in frequent bacteremia with oral organisms.70 Periodontal disease may predispose the patient to an increased incidence of bacteremia, including the presence of virulent gram-negative organisms associated with periodontitis. There is a greater risk of bacteremia after tooth brushing in patients with higher levels of plaque, calculus, and gingivitis as compared with those with minimal plaque and gingival inflammation.71 In fact, subjects with generalized gingival bleeding after brushing showed an almost eightfold increase in their incidence of bacteremia as compared with those with minimal gingival bleeding. An estimated 8% of all cases of infective endocarditis are associated with periodontal or dental disease, without a preceding dental procedure.32 The periodontium, when affected by periodontitis, also acts as a reservoir of endotoxins (LPSs) from gram-negative organisms. Endotoxins can pass readily into the systemic circulation during normal daily function, thereby inducing damage to the vascular endothelium and precipitating many negative cardiovascular effects. In a study of the incidence of endotoxemia after simple chewing, subjects with periodontitis were four times more likely to have endotoxin present in the bloodstream than subjects without periodontitis. Furthermore, the concentration of endotoxin present was more than fourfold greater in those with periodontitis as compared with healthy subjects.35
Thrombogenesis.
Platelet aggregation plays a major role in thrombogenesis, and most cases of acute MI are precipitated by thromboembolism. Oral organisms may be involved in coronary thrombogenesis. Platelets selectively bind some strains of Streptococcus sanguinis, which is a common component of supragingival plaque, and Porphyromonas gingivalis, which is a pathogen closely associated with periodontitis.44,45 The aggregation of platelets is induced by the platelet-aggregation–associated protein (PAAP) expressed on some strains of these bacteria.111 In animal models, the intravenous infusion of PAAP-positive bacterial strains resulted in alterations of heart rate, blood pressure, cardiac contractility, and electrocardiogram readings consistent with MI. Platelet accumulation also occurred in the lungs, which lead to tachypnea. No such changes were seen with the infusion of PAAP-negative strains. PAAP-positive bacteria caused aggregation of circulating platelets, which resulted in the formation of thromboemboli and the resultant cardiac and pulmonary changes. Thus, periodontitis-associated bacteremia with certain strains of S. sanguinis and P. gingivalis may promote acute thromboembolic events via interaction with circulating platelets.
Atherosclerosis.
Atherosclerosis is a focal thickening of the arterial intima, the innermost layer lining the vessel lumen, and the media, the thick layer under the intima that consists of smooth muscle, collagen, and elastic fibers (Figure 12-4).111 Early during the formation of atherosclerotic plaques, circulating monocytes adhere to the vascular endothelium. This adherence is mediated through several adhesion molecules on the endothelial cell surface, including intercellular adhesion molecule-1 (ICAM-1), endothelial leukocyte adhesion molecule-1 (ELAM-1), and vascular cell adhesion molecule-1 (VCAM-1).12,62 These adhesion molecules are upregulated by a number of factors, including bacterial LPSs, prostaglandins, and proinflammatory cytokines. After binding to the endothelial cell lining, monocytes penetrate the endothelium and migrate under the arterial intima. The monocytes ingest circulating low-density lipoprotein in its oxidized state and become engorged, thereby forming the foam cells that are characteristic of atheromatous plaques. After entering the arterial media, monocytes may also transform to macrophages. A host of proinflammatory cytokines such as interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), and prostaglandin-E2 (PGE2) are then produced, and these propagate the atheromatous lesion. Mitogenic factors such as fibroblast growth factor and platelet-derived growth factor stimulate smooth muscle and collagen proliferation within the media, thereby thickening the arterial wall.77 Atheromatous plaque formation and thickening of the vessel wall narrow the lumen and dramatically decrease blood flow through the vessel.111 Arterial thrombosis often occurs after an atheromatous plaque ruptures. Plaque rupture exposes circulating blood to arterial collagen and tissue factor from monocytes and macrophages that activate platelets and the coagulation pathway. Platelet and fibrin accumulation forms a thrombus that may occlude the vessel and result in ischemic events such as angina or MI. The thrombus may separate from the vessel wall and form an embolus, which may also occlude vessels, again leading to acute events such as MI or cerebral infarction (stroke).
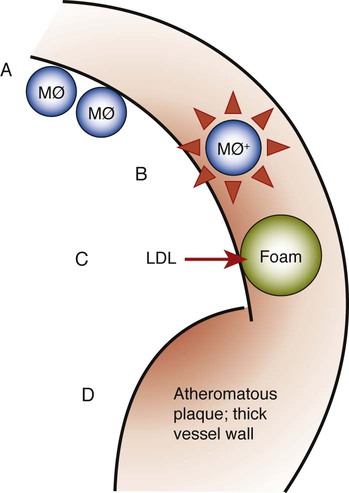
Role of Periodontal Disease in Myocardial or Cerebral Infarction
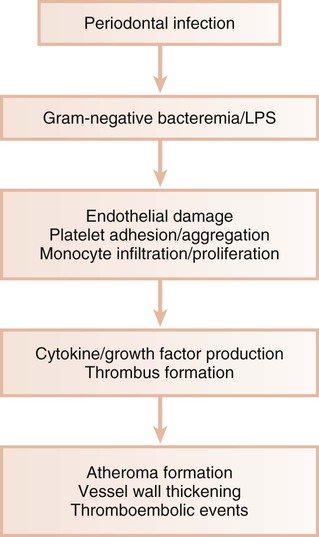
In animal models, gram-negative bacteria and associated LPS cause the infiltration of inflammatory cells into the arterial wall, the proliferation of arterial smooth muscle, and intravascular coagulation. These changes are identical to those seen with naturally occurring atheromatosis. Patients with periodontitis are at increased risk for the thickening of the walls of the major coronary arteries.10 In several studies of atheromas obtained from humans during endarterectomy, more than half of the lesions contained periodontal pathogens, and many atheromas contained multiple different periodontal species.18,42,140 Periodontal diseases result in chronic systemic exposure to products of these organisms. Low-level bacteremia may initiate host responses that alter coagulability, endothelial and vessel wall integrity, and platelet function, thereby resulting in atherogenic changes and possible thromboembolic events (Figure 12-5).
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


