CHAPTER 1 Pharmacodynamics
Mechanisms of Drug Action
DRUG-RECEPTOR INTERACTIONS
Receptor Classification
Besides enzymes (including coenzymes) and other easily solubilized proteins, at least two additional classes of receptors have been identified and are of clinical significance: nucleic acids and membrane-linked proteins. Nucleic acids serve as receptors for a limited number of agents. Certain antibiotics and antineoplastic compounds interfere with replication, transcription, or translation of genetic material by binding, sometimes irreversibly, to the nucleic acids involved. Other drugs, including thyroid hormones, vitamin D analogues, sex steroids, and adrenal corticosteroids, also modify transcription, but here the affected DNA becomes activated or inhibited as a consequence of drug interaction with a separate receptor protein in the cytosol or nucleus of the cell, as described subsequently. The most common receptors of drugs are those located on or within the various membranes of the cell. Their study has been greatly aided in recent years by developments in genomics, proteomics, and informatics. Membrane transporter proteins and metabolic enzymes, described in Chapter 2 for their influence on drug disposition, are themselves targets of drug action. Of greater significance are the many integral membrane proteins that function as receptors for endogenous regulatory ligands, such as neurotransmitters, hormones, and other signaling molecules.
Receptors involved in physiologic regulation can be grouped by molecular structure and functional characteristics into several superfamilies. Most of these receptors have one or more extracellular ligand-binding domains linked by one or more lipophilic membrane-spanning segments to an effector domain often, but not always, located on the cytoplasmic side of the membrane. This arrangement is ideal for the translation of an extracellular signal into an intracellular response. Usually, the endogenous ligand “signal” is hydrophilic and incapable of passive diffusion through the cell membrane. For lipophilic regulatory ligands, such as for thyroid hormone and various steroids, a separate superfamily of intracellular receptors exists. Commonly, drug binding exposes a DNA-binding site on the receptor protein, allowing the receptor to interact with DNA and alter transcription. These major classes of receptors are illustrated in Figure 1-1 and described subsequently.
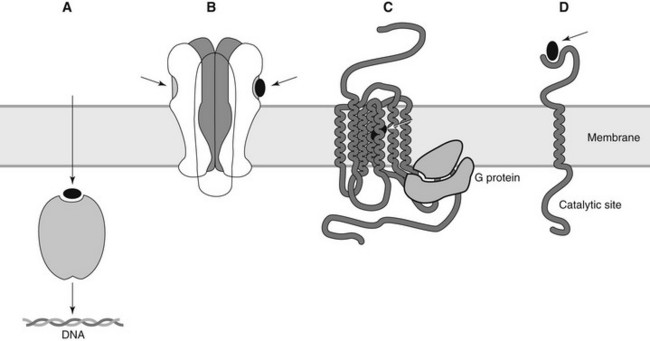
Ion channel–linked receptors
There are two general classes of ion channels: voltage gated and ligand gated. Voltage-gated ion channels are activated by alterations in membrane voltage. Voltage-gated Na+ channels open when the membrane is depolarized to a threshold potential and contribute to further membrane depolarization by allowing Na+ influx into the cell. As described in Chapter 16, local anesthetics such as lidocaine bind to voltage-gated Na+ channels, leading to blockade of neuronal depolarization. Specific voltage-gated ion channels also exist for K+, Ca++, H+, and Cl−.
The nicotinic receptor (Figure 1-2), the first membrane-bound drug receptor to be fully characterized,12,22 is an important example of a ligand-gated ion channel. An oligomeric structure, the polypeptide constituents of the nicotinic receptor are arranged concentrically to form a channel through which small ions can traverse the plasma membrane when the receptor is activated by the binding of two acetylcholine (ACh) molecules. As is the case with other ion channels, numerous subtypes of nicotinic receptors exist expressing differing affinities for specific ligands.
G protein–linked receptors
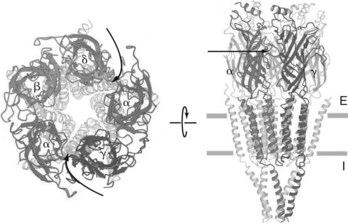
G protein–linked receptors, sometimes referred to as metabotropic receptors, constitute the largest superfamily of integral membrane proteins, and collectively serve as targets for approximately half of all nonantimicrobial prescription drugs.9,11 The basic structure of these receptors includes a common seven-membered transmembrane domain. Generally, metabotropic receptors greatly amplify extracellular biologic signals because they activate G proteins, which activate ion channels or, more commonly, other enzymes (e.g., adenylyl cyclase), leading to the introduction or formation of a host of internal second messengers for each extracellular signal molecule detected. This amplification system, which also usually involves an extended duration of activation of G proteins relative to the binding of drug to the receptor, may explain why maximal pharmacologic effects are often observed when only a small proportion of receptors are activated.
G proteins are heterotrimers consisting of α, β, and γ subunits. After receptor activation, guanosine diphosphate attached to the α subunit is replaced by guanosine triphosphate, and the heterotrimer splits into the α monomer and βγ dimer. Many, but not all, of the observed cellular actions are caused by the α subunit (see Figure 5-7). Gαs, the specific α subunit for the G protein associated with β-adrenergic receptors, activates adenylyl cyclase, which catalyzes the synthesis of cyclic adenosine 3′,5′-monophosphate (cAMP).9 cAMP activates protein kinase A, which catalyzes the phosphorylation of serine and threonine residues of certain intracellular proteins, leading to altered cellular function.
The G protein system is complex and still incompletely understood. One receptor subtype may activate different G proteins, several receptor subtypes may activate the same G protein, and the ultimate target proteins can exist in tissue-specific isoforms with differing susceptibilities to secondary effector systems. The different G protein pathways can also interact with one another. The complexity of G protein signal transduction provides a sophisticated regulatory system by which cellular responses can vary, depending on the combination of receptors activated and the cell-specific expression of distinct regulatory and target proteins. Several specific membrane-bound G proteins are discussed in Chapter 5.
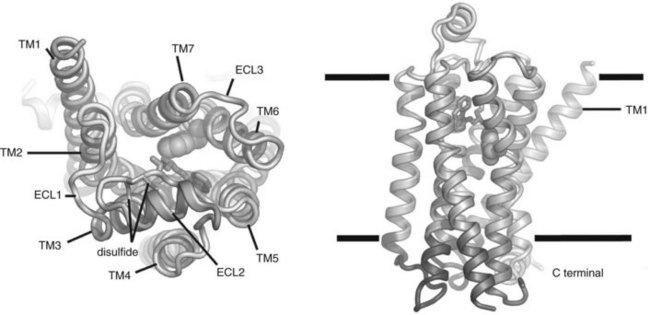
Figure 1-3 depicts the structure of the mammalian β2 receptor based on x-ray crystallography studies and how it is believed to be arranged within the plasma membrane.11
Enzyme-linked receptors
Many forms of cancer seem to involve mutant variants of enzyme-linked receptors in which the catalytic site or associated nonreceptor protein kinase is continuously activated.4 Approximately half of all oncogenes discovered to date encode for continuously activated protein kinases.
Intracellular receptors
Lipophilic substances capable of crossing the plasma membrane may activate intracellular receptors. Sex steroids, mineralocorticoids, glucocorticoids, thyroid hormones, and vitamin D derivatives all activate specific nuclear receptors that influence DNA transcription.8,20 The typical nuclear receptor is composed of three major subunits: the carboxyl end of the receptor forms the ligand-binding domain, the adjacent segment includes the DNA-binding region, and the amino terminus constitutes the transcription-modulating domain. When a drug (or hormone) binds to the receptor, it folds into the active configuration and dimerizes with a partner receptor. The conformational change results in a dramatic increase in binding to specific DNA sequences. Binding of thyroid hormone to its receptor produces more than a 10-fold increase in receptor affinity for binding to DNA.20 DNA binding of the activated receptor often initiates transcription, leading to increased production of specific proteins. Because this type of signal transduction requires protein synthesis, drugs that activate intracellular receptors typically have a delay of several hours before the onset of their pharmacologic effect. (For this reason, glucocorticoids cannot be used as primary drugs for the management of anaphylaxis.) In some systems, the binding of the drug-receptor complex inhibits transcription. Regardless of the specific mechanism involved, however, the intensity and duration of drug effect is temporally independent of the plasma concentration.
Drug-Binding Forces
Implicit in the interaction of a drug with its receptor is the chemical binding of that drug to one or more specific sites on the receptor molecule. Five basic types of binding may be involved (Figure 1-4).
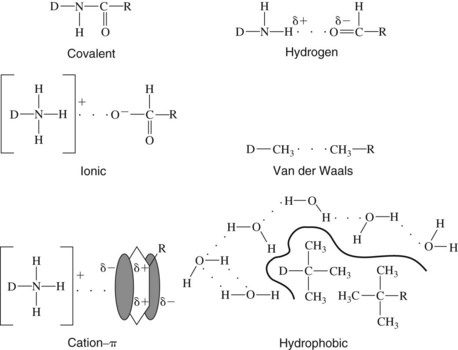
FIGURE 1-4 Major chemical bonds associated with drug-receptor interactions, where D is drug and R is receptor.
Covalent bonds
Covalent bonds arise from the sharing of electrons by a pair of atoms. Although covalent bonds are required for the structural integrity of molecules, they are generally not involved in drug-receptor interactions. Most drugs reversibly associate with their receptors. As described in Chapter 2, the duration of action of these agents is related to how long an effective drug concentration remains in the vicinity of the drug receptors. This time may vary from a few minutes to many days, but usually is on the order of several hours. With bond energies of 250 to 500 kJ/mol, the stabilities of covalent linkages are so great that, when formed, drug-receptor complexes are often irreversible. In these instances, the duration of action is not influenced by the concentration of unbound drug surrounding the receptors. Instead, it may depend on the synthesis of new receptors or on the turnover of the affected cells, processes that often take days to weeks. When the receptors happen to compose or influence the genetic material of a cell, drug effects may be permanent.
Cation-π interactions
Although benzene and similar aromatic compounds are hydrophobic solvents, their π electron clouds are capable of interacting with positively charged ions.5 Phenylalanine, tyrosine, and tryptophan—amino acids with aromatic side groups—retain this ability. These amino acids are common constituents at receptor sites for such positively charged drugs as ACh, dopamine, epinephrine, and 5-HT. Individual bond energies are similar to those of hydrogen bonds described subsequently; however, interactions between multiple aromatic amino acids and a single cationic moiety commonly strengthen the overall interaction.
Structure-Activity Relationships
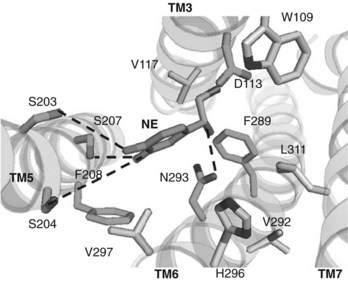
A recent example of SARs is provided by the study of the binding of norepinephrine and related drugs to the β2-adrenergic receptor (Figure 1-5).2 The norepinephrine molecule is composed of a catechol residue (a benzene ring with two hydroxyl groups in the meta and para positions) connected by a two-carbon intermediate chain to a nitrogen terminus that is positively charged at physiologic pH. The presence of a cationic nitrogen locus is essential for full activity; loss of the ionic charge by removing the nitrogen moiety or replacing it with a nonionic carbon group virtually eliminates drug action, as does replacement of the receptor’s aspartate residue (D113) with a neutral amino acid. Hydrogen bonds involving both ring hydroxyl groups with corresponding serine residues (S203, S204, and S207) greatly increase potency (by 25-fold, 33-fold, and 39-fold, respectively) but preclude entry of the drug into the central nervous system (CNS). Replacement of a hydroxyl with a larger group generally eliminates agonist activity at β receptors but may result in antagonistic effects. Another hydrogen bond between the β-hydroxyl group and its asparagine counterpart (N293) increases binding affinity 38 times. The distance separating the catechol and nitrogen moieties of the molecule is likewise crucial for full activity. Electrostatic interactions involving the benzene ring and aromatic amino acid residues of the receptor protein (e.g., F290) also contribute to the binding of norepinephrine.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


