Fig. 5.1
HGF/SF and MET structures. Domain structures of HGF/SF and MET. HGF/SF consists of an N-terminal domain, four kringle domains, and a serine protease homology motif. MET includes extracellular SEMA, PSI, and IPT domains and an intracellular juxtamembrane domain, a Kinase domain, and C-terminal docking sites. The arrangement and sequence of the substrate-binding sites of MET are shown
Two distinct activation modalities responsible for the processing of pro-HGF/SF to mature HGF/SF have been proposed. One is cleavage by the soluble serum serine protease HGFA, which also requires endoproteolytic processing by serine proteases such as thrombin to become active [39, 43, 44]. During tissue repair, the active form of HGFA binds to heparin molecules to ensure the localized activation of HGF/SF at the injury site [45]. Other extracellular proteases such as blood coagulation factor XIIa, tissue type plasminogen activator, and urokinase are also known to be able to cleave pro-HGF/SF to form mature HGF/SF [40]. The second activation modality involves membrane-anchored serine proteases, such as matriptase and hepsin that are expressed on the surface of MET-expressing target cells [17, 46]. These soluble or membrane-anchored HGF/SF-activating proteases are precisely regulated by the cellular serine protease inhibitors HGF/SF activator inhibitor 1 (HAI1, also known as SPINT1) and HAI2 (also known as SPINT2) [47, 48]. The inflammatory environment and tumor stroma are known to overexpress proteases that are involved in pro-HGF activation while limiting expression of protease inhibitors [18]. In HNSCC tumors, the oncogenic protease matriptase is ubiquitously co-expressed with MET and has been reported to cleave and convert the inactive single-chain HGF/SF into the functional double-chain HGF/SF thereby activating MET to initiate the signaling pathways leading to carcinogenesis, invasion, and metastasis in vitro and in vivo [49]. On the contrary, the attenuation of HAI1 and/or HAI2 expression promotes invasive growth and metastasis [17, 50]. Therefore, detailed knowledge of this combined transcriptional and posttranslational regulation of HGF/SF, which leads to optimal MET activation on target cells, is essential in understanding the pathophysiological roles of HGF/SF and for developing novel therapeutic intervention.
Identification and Activation of MET
MET is a member of the receptor tyrosine kinase (RTK) family and is encoded by the MET proto-oncogene located on chromosome 7q21-31 [51]. MET was initially discovered as an oncogene in osteogenic sarcoma cells and was later identified as the receptor for HGF/SF [52, 53]. In osteosarcoma, MET was discovered to be involved in a chromosomal rearrangement in which its tyrosine kinase domain was fused to translocating promoter region (TPR) [52]. This MET-TPR rearrangement results in the constitutive activation of MET [52]. Expression of TPR-MET in transgenic mice promotes the development of multiple epithelial-derived tumors [54]. Additional studies reveal that HGF/SF and MET play critical roles in the normal development of the placenta and liver, tissue repair, morphogenic differentiation, organization of three-dimensional tubular structures, cell growth and angiogenesis, epithelial-mesenchymal transition (EMT) of the epithelial dermomyotome, migration of myogenic precursor cells, as well as tumorigenesis and malignant progression of various tumors [17, 55–57]. MET signaling is also required for tooth development, myogenesis in the developing tongue, wound healing, and gingival tissue remodeling in the oral cavity [57, 58]. MET is mainly expressed in epithelial cells but is also found in endothelial cells, myoblasts, spinal neurons, and hematopoietic system cells [18].
The mature MET receptor is formed by proteolytic processing of a native MET protein to a disulfide-linked single-pass heterodimer composed of a 50 kDa extracellular α-subunit and a 145 kDa transmembrane β-subunit (see Fig. 5.1) [16, 34]. The extracellular portion of MET is composed of three domains: sema, plexin-semaphorin-integrin (PSI), and immunoglobulin-plexin-transcription (IPT) domains. The sema domain spans the N-terminal 500 residues and encompasses the entire α-subunit and part of the β-subunit [16]. The sema domain of MET interacts with the low-affinity site of the HGF β-chain [42] and has been shown to promote MET dimerization and activation [59]. The PSI domain of MET spans approximately 50 residues and includes four disulfide bonds [16]. Adjacent to the PSI domain, 400 residues of four IPT subdomains form the transmembrane helix and connect with the intercellular portion of MET [16]. The high-affinity site of the HGF α-chain interacts with the IPT3 and IPT4 subdomains [41]. The intracellular segment of MET is composed of a juxtamembrane domain, a catalytic kinase domain, and a multifunctional docking site at the C-terminal. Following HGF binding to the extracellular domains of MET, the kinase activity of MET is initiated through receptor dimerization and transphosphorylation of four major tyrosines, Tyr1234, Tyr1235, Tyr1349, and Tyr1356 (also known as Y1234, Y1235, Y1349, and Y1356), within the intracellular β-subunit segment of MET [16, 51]. Tyrosine kinases within the kinase domain autophosphorylate Tyr1234 and Tyr1235 upon MET receptor dimerization, leading to a subsequent transphosphorylation of Tyr1349 and Tyr1356 in the carboxy-terminal docking site [6, 16, 17, 51]. Phosphorylation of Tyr1349 and Tyr1356 recruits numerous Src homology 2 (SH2) domain-containing scaffolding adaptors including the growth factor receptor-bound protein 2 (GRB2), c-Src, Src homology and collagen protein (Shc), the transcription factor signal transducer and activator of transcription 3 (STAT3), GRB2-associated-binding protein 1 (GAB1), and phospholipase Cγ (PLCγ) [16, 51, 60, 61]. These scaffolding molecules are phosphorylated on tyrosine residues and provide additional levels of signal branching by attracting more adaptor molecules and enzymes such as phosphoinositide 3-kinase (PI3K), tyrosine-protein phosphatase non-receptor type 11 (PTPN11, also known as SHP2), Src homology 2 domain-containing tyrosine phosphatase (SHP), Ras-related C3 botulinum toxin substrate 1 (RAC1), p21-activated kinase (PAK), focal adhesion kinase (FAK), and others [17, 60, 62, 63]. GAB1 is a unique multi-adaptor protein associated with MET. Once bound to and phosphorylated by MET, GAB1 provides binding sites for more downstream adaptors [64]. GAB1 can bind to MET directly or indirectly through GRB2 [51]. Sustained phosphorylation of GAB1 in response to HGF/SF results in attenuation of the morphogenic invasive response [65] and also leads to the inhibition of several GAB1-dependent signaling pathways [34]. Genetic experiments in mice have demonstrated that the interaction between GAB1 and different effector molecules results in distinct biological roles [16, 66]. For example, knock-in mice that carry point mutations in the PI3K-binding sites of GAB1 affects embryonic eyelid closure and keratinocyte migration during development; however, point mutations in SHP2-binding sites of GAB1 interrupt placental development and muscle progenitor cell migration to the limbs [66]. Moreover, phosphorylation of serine 985 in the juxtamembrane domain of MET is known to downregulate the kinase activity of MET [67]. Taken together, this unique MET-dependent signaling apparatus that utilizes multifunctional scaffolding adaptors leads to efficient activation of downstream biochemical pathways and regulation of gene expression.
Activation of MET-dependent signaling pathways (illustrated in Fig. 5.2) controls numerous cellular responses such as cell growth, proliferation, transformation, survival, and migration. One of the major downstream pathways triggered by MET activation is the mitogen-activated protein kinase (MAPK) signaling pathway [16, 51, 68]. This distinctive signaling cascade involves the activation of a series of three protein kinases: MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK), and MAPK [16, 69]. Extracellular signal-regulated kinases 1 and 2 (ERK 1/2) are MAPKs that are mainly activated by tyrosine kinase-dependent stimulation of Ras GTPase [70, 71]. Ras proteins are cell membrane-bound hydrolase enzymes that can bind to and hydrolyze guanosine triphosphate (GTP). The Ras GTPase proto-oncogene is found to be mutated in a high percentage of human tumors including HNSCC and leads to the activation of signaling pathways that promote tumor growth and metastasis [15]. Upstream of Ras and GRB2 can either directly interact with the C-terminal multifunctional docking site of MET or indirectly bind to the Shc adaptor protein associated with MET [60, 72]. Upon GRB2 interaction, the Ras guanine nucleotide exchanger factor (GEF) son of sevenless (SOS) can be recruited to form a GRB2-SOS complex or Shc-GRB2-SOS complex to activate Ras [16, 51]. The active GTP-bound Ras binds to the serine/threonine kinase v-raf murine sarcoma viral oncogene homolog B1 (Raf), which acts as MAPKKK in the MAPK pathway. Raf kinase then phosphorylates MAPK/ERK kinase 1 (MEK1, also known as MAPKK1) or MEK2 (also known as MAPKK2) [16, 68]. Consequently, MEK1 and MEK2 phosphorylate ERK1/2, the effectors of the MAPK cascade [68, 71]. Phosphorylated ERK1/2 activates ETS domain-containing protein (Elk1), to activate several transcription factors, such as v-ets avian erythroblastosis virus E26 oncogene homology 1 (ETS1), and regulates gene transcription to promote cell proliferation, cell motility, transformation, differentiation, and cell cycle progression [16, 73]. ERK1/2 also promotes the transcriptional upregulation and dimerization of c-Jun and c-Fos to form activator protein 1 (AP-1). AP-1 was one of the first mammalian transcription factors identified and is known to regulate a wide range of cellular processes, including cell proliferation, survival, differentiation, invasion, and metastasis [74, 75]. AP-1 is composed of dimeric basic region-leucine zipper (bZIP) proteins that belong to the Jun (c-Jun, JunB, JunD), Fos (c-Fos, FosB, Fra-1, and Fra-2), v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog (MAF), and activating transcription factor (ATF) subfamilies [74].
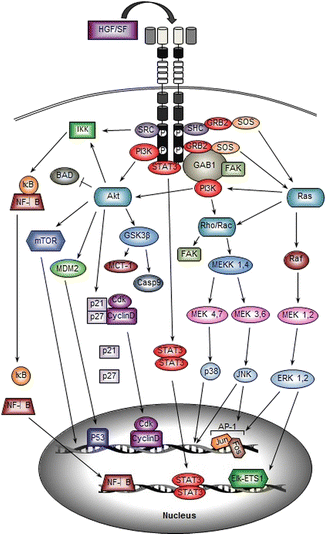
Fig. 5.2
HGF/SF-MET signal transduction. The signaling molecules and major downstream pathways associated with HGF/SF-induced MET activation are illustrated. A scaffolding adaptor such as GAB1 is recruited directly and/or indirectly to activate MET, which results in activation of intracellular signaling molecules such as PI3K and Ras. The signal transduction cascades lead to activation of multiple transcription factors, promoting gene amplification and cellular responses
The PI3K signaling pathway can also be activated directly by MET or indirectly by MET-activated Ras [16]. The Ras-PI3K complex is able to activate other MAPK family members known as c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38). Activation of JNK and p38 occurs by stimulation of the small GTP-binding proteins Rac and Rho, which are members of the Ras GTPase superfamily [76]. Ras-PI3K-stimulated Rac can activate MEK kinase 1 (MEKK1) and MEKK4, the first MAPKKK components of the MAPK cascade, which subsequently phosphorylates MEK4 and MEK7 to activated JNK1, JNK2, and JNK3 [16]. On the other hand, MEKK1 and MEKK4 can also phosphorylate MEK3 and MEK6 to activate p38α, p38β, p38γ, and p38δ [76, 77]. The JNKs and p38 regulate various cellular processes including cell proliferation, motility, differentiation, transformation, and apoptosis. Several studies have demonstrated that cytoskeletal regulation and focal adhesion assembly also depends on Rho activity [78]. In human oral squamous cell carcinoma (OSCC), inhibition of Rho using clostridium botulinum C3 exoenzyme (C3) significantly reduces the motility of OSCC cells and decreases the tyrosine phosphorylation of focal adhesion kinase (FAK) [79]. FAK can directly interact with SH2-containing proteins to get phosphorylated or can be indirectly activated by Rac-/Rho-MAPK pathway [80]. FAK has been shown to play a key role in both normal and tumor cell migration [80].
PI3K activation also leads to the formation of phosphatidylinositol-3,4,5-triphosphate (PtdIns(3,4,5)P3 or PIP3) thereby recruiting pleckstrin homology domain (PH domain)-containing molecules such as protein kinase B (PKB, also known as Akt) [16, 81]. PIP3 binding to the PH domain of Akt results in a conformational change that allows phosphoinositide-dependent protein kinase 1 (PDK1) to phosphorylate threonine 308 within the Akt catalytic domain [82]. Activated Akt promotes cell survival in multiple ways. First, Akt can phosphorylate and inactivate the pro-apoptotic protein BCL-2 antagonist of cell death (BAD). Second, Akt can activate the E3 ubiquitin-protein ligase mouse double minute 2 homology (MDM2) [16]. MDM2 is involved in stabilizing tumor protein p53 (TP53, also known as p53). Akt-mediated phosphorylation of MDM2 at serine 186 increases MDM2 ubiquitination of p53, which results in degradation of the p53 protein [83, 84]. Moreover, Akt directly phosphorylates glycogen synthase kinase 3β (GSK3β) in a highly conserved N-terminal regulatory site [85]. This phosphorylation inactivates GSK3β which results in increased expression of the anti-apoptotic Bcl-2 family member myeloid leukemia cell differentiation protein (MCL-1), caspase 9, and other positive cell cycle regulators such as Myc and cyclin D1 [16, 85]. Active Akt also promotes phosphorylation of cyclin-dependent kinase inhibitors p21CIP1/WAF1 and p27KIP1, resulting in their degradation in the cytoplasm, to allow nuclear translocation of the cyclin-dependent kinase and cyclin D complex (CDK-CyclinD), thereby promoting cell proliferation [86, 87]. The PI3K-Akt pathway also activates mammalian target of rapamycin complex 1 (mTORC1) by phosphorylating the proline-rich Akt substrate of 40 kDa (PRAS40) to relieve the PRAS40-mediated inhibition of mTORC1 [88]. At the same time, PI3K-Akt phosphorylates mTORC1-negative regulator the tuberous sclerosis complex 2 (TSC2, also known as tuberin) to allow inactivation of TSC2 [89]. Activation of mTORC1 stimulates protein synthesis and physical cell enlargement [16].
Transcription factors such as nuclear factor kappa light chain enhancer of activated B cells (NF-κB) are also activated through the MET-PI3K-Akt pathway leading to the induction of mitogenic and anti-apoptotic gene expression [90–93]. Activation of the canonical NF-κB pathway is initiated by the signal-induced degradation of the inhibitors of κB (IκBs) bound to NF-κB in the cytoplasm by the IκB kinase (IKK) [90]. PI3K-Akt and SRC act as signaling intermediates to mediate IKK activation in the cytoplasm [93]. Currently, the direct activator of IKK remains unknown [93]. Upon activation, IKK phosphorylates serine 32 and serine 36 in the regulatory domain of IκB [90, 91]. Once phosphorylated, IκB is targeted for ubiquitination and subsequent degradation, leading to the release of NF-κB [90]. NF-κB then translocates to the nucleus to stimulate the transcription of various genes [92]. In HNSCC, NF-κB can induce the transcription of growth-regulated oncogene-1 (GRO-1), a neutrophil chemoattractant that is linked to HNSCC angiogenesis and lymph node metastasis [94].
Other important downstream molecules of HGF/SF-MET signaling are the STAT proteins. STAT3, in particular, directly binds to a transphosphorylated docking site of MET and becomes activated when it is subsequently phosphorylated by MET [64, 95]. Phosphorylated STAT3 dissociates from the MET receptor and homodimerizes through its SH2 domain and consequently translocates to the nucleus to regulate the expression of genes involved in cell proliferation, differentiation, and invasion [16, 96]. Recent studies reveal that STAT3 and MCL-1 interact with each other and can promote apoptosis, mesenchymal to epithelial transition, and metastasis [97]. Constitutively, activated STAT3 also results in increased tumor cell proliferation, survival, and invasion by suppressing antitumor immunity while promoting tumor-promoting inflammation [96, 97]. In OSCC, immunohistochemical examination of 48 patient samples reveals a significant correlation between MET expression, the constitutive activation of STAT3, and tumor stage, indicating that activation of STAT3 by MET is associated with the malignant progression of OSCC [98].
5.3 Regulation of HGF/SF-MET Signaling
The number of cell surface proteins that play a role in MET signaling is growing constantly. Understanding the significance of MET cross talk with these molecules and the positive and negative regulation of MET will provide valuable information for the selection of appropriate treatment strategies in HNSCC patients.
Positive Regulation
CD44 is a transmembrane glycoprotein involved in cell to cell interaction, cell adhesion between the extracellular matrix and the intracellular actin cytoskeleton, and migration [6]. Various isoforms of CD44 exist (CD44v1-CD44v10), which differ in their extracellular domain and are produced by alternative splicing of RNA [99]. Among them, the CD44 isoform containing the v6 sequence (CD44v6) cooperates with binding of HGF/SF to MET to activate HGF/SF-MET signaling in normal and transformed epithelial cells [16, 99]. Interestingly, in vivo experiments have demonstrated that MET is haploinsufficient in CD44 −/− mice [100]. Upon MET activation, the intracellular domain of CD44v6 associates with MET to recruit GRB2; F-actin; the ezrin, radixin, and moesin (ERM) family of proteins; and SOS [99, 101]. Consequently, the GRB2-ERM-SOS complex promotes efficient activation of Ras [101]. Thus, CD44 functions as a co-receptor to HGF/SF-MET and amplifies MET signaling by regulating its intracellular structure and topography rather than by directly interacting with effector molecules. When CD44v6 is absent, intercellular adhesion molecule-1 (ICAM-1) can substitute for CD44v6 as a co-receptor for MET signaling to provide similar results [102]. In endothelial cells, CD44v10, another CD44 isoform, also acts as a co-receptor to facilitate MET signaling [103].
The α6β4 integrin receptor is involved in normal keratinocyte migration, tumorigenesis, and invasive growth [6, 104, 105]. The α6β4 receptor also has a scaffolding function similar to GAB1 and acts as an adaptor protein by associating with MET [16]. MET and α6β4 interaction intensifies MET signaling by providing supplementary docking platforms for the localized recruitment of additional signal transducers [106]. The β-subunit of the α6β4 receptor directly binds to MET and becomes phosphorylated at three tyrosine residues on the cytoplasmic domain, which in turn provides extra binding sites for SHC, PI3K, and SHP2 [16, 105]. In addition, GRB2 can interact with α6β4-bound SHC to intensify the MET signal [105, 107], and α6β4-bound SHC can also activate Ras-MAPK pathway to further potentiate activation of MET downstream pathways [108]. In HNSCC, α6β4 expression is stronger in metastatic HNSCC cell lines compared to nonmetastatic cells [109]. Further, HNSCC tumor tissues also show elevated levels of α6β4 expression localized at the invasive fronts [110].
Other RTKs are also known to positively interact with MET to activate MET-dependent pathways and enhance MET-mediated biological functions. Investigating the cross talk between MET and other RTKs in great depth is important so that the mechanisms of chemoresistance can be better understood. The extracellular sema domain of MET shares structural homology with a family of semaphorin receptors such as plexins and neuropilins and forms a seven-bladed beta-propeller that allows multiple interactions between MET and plexins [16, 111, 112]. These semaphorin receptors are able to transactivate MET in an HGF-independent manner, leading to activation of the MET signaling pathway and resulting in biological responses such as modification of the cytoskeleton [112–114]. In HNSCC, semaphorin 4D (SEMA4D) and neuropilin-1 are upregulated and have been shown to be associated with poor prognosis and promote angiogenesis, cell migration, and invasion [115, 116]. Numerous studies also reveal that ligand activation of EGFR family members can transactivate MET in cells that express both MET and EGFR in the absence of HGF/SF [117]. EGFR can bind to MET and form a heterodimeric receptor complex, activating both tyrosine kinases through transphosphorylation. Introducing epithelial growth factor (EGF) or TGFα to cells expressing both MET and EGFR results in phosphorylation of MET and activation of both EGFR and MET signal cascades [118, 119]. The mechanism of cross talk between these two pathways involves the Ras-MAPK pathway and associated molecules to stimulate each other [120]. EGFR transactivation of MET through the Ras-MAPK pathway stimulates the production of tissue inhibitor of metalloproteinase 3 (TIMP3), which cleaves the extracellular domain of MET [121]. This truncated MET protein promotes proliferation and cell transformation [122]. MET can also activate EGFR through Ras-MAPK-dependent transcription of EGF [123]. EGF is then secreted to the ECM to bind with EGFR in an autocrine or paracrine manner [123].
Similarly, cross talk between MET and both the tyrosine-protein kinase receptor UFO (AXL) and platelet-derived growth factor receptor (PDGFR) RTKs has been found to play a role in bladder cancer [51, 120]. AXL is a member of the Tyro-3, Axl, and Mer (TAM) RTK family [124]. AXL can stimulate cell proliferation, survival, and migration through the MEK/ERK MAPK signaling pathway [125]. PDGFR signaling is involved in tissue development, EMT during metamorphic skin remodeling, and mesenchymal cell migration and proliferation [126]. MET is frequently co-expressed with AXL and PDGFR, and overexpression of AXL and PDGFR has been detected in various human cancers and is associated with invasiveness and/or metastasis of carcinoma of the breast, kidney, and bladder [127–129]. Interaction of MET with the recepteur d’origine nantais (RON) receptor, a MET RTK subfamily member, has also been shown to cause transphosphorylation of the MET receptor in the absence of HGF/SF [6, 51, 130]. MET directly interacts with RON to form a heterodimer. The MET/RON heterodimer transphosphorylates the catalytic region of MET at Tyr 1234 and Tyr 1235 and RON at Tyr 1238 and Tyr 1239, respectively, thereby generating signal transducer docking sites at Tyr 1349 and Tyr 1356 on MET and Tyr 1353 and Tyr 1360 on RON for downstream signaling molecules [130]. In addition, G-protein-coupled receptor (GPCR) agonists including bradykinin and thrombin can activate MET signaling through a reactive oxygen species-dependent mechanism [131].
Negative Regulation
The most common negative regulatory mechanisms of RTKs such as MET involve protein-tyrosine phosphatases (PTPs). As previously discussed, the function of MET depends on phosphorylation of intercellular tyrosines. A sophisticated balance between intrinsic tyrosine kinase activity of MET and counteracting PTP activity determines net tyrosine phosphorylation of MET, which governs the outcome of MET function in the cell. Several PTPs have been shown to regulate MET tyrosine phosphorylation including density-enhanced phosphatase-1 (DEP-1, also known as RPTP-J), leukocyte common antigen-related receptor protein-tyrosine phosphatase (LAR-RPTP, also known as PTPRF), receptor protein-tyrosine phosphatase β (RPTP-β), protein-tyrosine phosphatase 1B (PTP1B, also known as PTPN1), and T cell protein-tyrosine phosphatase (TCPTP, also known as PTPN2) [6, 16, 51]. DEP-1, LAR-RPTP, and RPTP-β are receptor type PTPs. DEP-1 preferentially dephosphorylates the GAB1-binding site Tyr 1349 and a C-terminal Tyr 1365 [132]. RPTP-β also specifically dephosphorylates MET at Tyr 1356 and inhibits downstream MEK 1,2 and ERK activation [133–136]. Abolition of endogenous RPTP-β expression increases basal and HGF/SF-stimulated MET phosphorylation at Tyr 1356 in primary human keratinocytes [133]. Conversely, expression of RPTP-β in human keratinocytes reduces HGF-mediated angiogenesis, proliferation, and motility [133]. In HNSCC, MET function also depends on tyrosine phosphorylation that is regulated by RPTP-β. Knockdown of endogenous RPTP-β in HNSCC cells from primary tumors results in the accumulation of MET tyrosine phosphorylation and activation of MAPK signaling [135]. In addition, RPTP-β expression is significantly downregulated in HNSCC cells derived from metastatic tumors compared to nonmetastatic [135]. This study also reports that six out of eight HNSCC tumors have reduced levels of RPTP-β expression in comparison with normal oral tissues, indicating the importance of RPTP-β function to regulate MET activation in tumor biology of HNSCC [135]. Non-receptor types PTP1B and TCPTP have been shown to dephosphorylate Tyr 1234 and Tyr 1235 on the kinase domain of MET [6, 16]. The ability of PTPs to dephosphorylate specific tyrosine residues that are required for MET-induced signaling suggest that PTPs may function in regulating the specificity of signals induced by MET, rather than acting as a simple “off switch” to inhibit MET activity.
Ligand or ubiquitination-independent proteolysis of MET provides a low-grade reduction of MET signaling under steady-state conditions [16]. The first step is mediated by a disintegrin and metalloprotease (ADAM), which cleaves and releases the extracellular domain of MET, generating a soluble N-terminal fragment and a membrane-associated cytoplasmic tail [137]. This process is known as shedding. The extracellular shedding of MET not only decreases the available number of MET receptors on the cell surface but also introduces a decoy moiety that can directly interact with both HGF/SF and MET to further inhibit MET signaling [138, 139]. Secondly, the membrane-bound cytoplasmic remnant of MET gets further proteolyzed by γ-secretase, which produces a labile intracellular fragment subject to proteasome-mediated degradation [137].
Negative regulation through endocytosis of the MET receptor and desensitization of MET signaling has also been extensively studied and reveals the paradoxical effect of MET internalization on HGF/SF-MET-mediated signal transduction. Phosphorylation of Tyr 1003 in the juxtamembrane domain of HGF/SF-activated MEt allows binding of casitas B-lineage lymphoma (CBL), an E3 ubiquitin-protein ligase [16, 140]. This interaction results in monoubiquitylation of MET at multiple sites, attracting endocytic adaptors with ubiquitin-binding domains and sorting into clathrin-coated areas on the plasma membrane [141]. Through an endosomal network, MET accumulates in the internal membranes of multivesicular bodies of the endosomal compartment and in the Golgi apparatus, resulting in termination of MET signaling and MET recycling from the plasma membrane [140]. Finally, the multivesicular bodies fuse with the lysosomes and allow lysosomal degradation of MET [141]. In addition, MET-CBL interaction attracts CIN85-endophilin complex, which can cause an inner leaflet of the plasma-membrane bilayer to adapt invagination curvature and scission to promote endocytosis [142, 143]. Interestingly, even though MET endocytosis negatively regulates MET signaling, recent studies reveal that MET internalization can also promote HGF/SF-MET signaling. Protein kinase Cε (PKCε) and protein kinase Cα (PKCα) are key regulators of MET internalization and relocation of MET to an endosomal compartment by stimulating the endosomal network [16, 144]. PKCε can also stimulate internalized MET to sustain activation of ERK1/2, thereby triggering relocation of ERK1/2 to the front edge of the cell where the focal adhesion complex accumulates [145–147]. Knockdown of PKCε blocks the recruitment of ERK 1/2 to focal adhesions and negatively impacts HGF-mediated cell migration [145, 146]. Further, PKCα-mediated endocytosis of MET provides protection for MET-triggered phosphorylation of STAT3 inside the cytoplasm against phosphatase activity and grants nuclear translocation of active STAT3 [144, 145, 147].
HGF/SF-independent regulation of MET signaling is also accomplished by binding of MET to the transmembrane molecule known as leucine-rich repeats and immunoglobulin (Ig)-like domain 1 (LRIG1). LRIG1 destabilizes the MET receptor and impairs its ability to respond to HGF/SF. LRIG1 knockdown increases MET receptor half-life, while overexpression of LRIG1 impairs HGF/SF-MET signaling, indicating an essential negative regulatory role of LRIG1 in MET activation [148]. Other studies show that phosphorylation of Tyr 221 on adapter molecule crk II (CRKII, also known as proto-oncogene c-Crk) by the Abelson murine leukemia viral oncogene homolog (ABL) tyrosine kinase creates a negative feedback loop to regulate motile responses upon MET stimulation by HGF/SF [149]. Increased intracellular calcium levels also have inhibitory effects on MET signaling via a PKC-independent mechanism. In vivo labeling studies with radioactive orthophosphate have shown that increased intracellular calcium induces Ser145 phosphorylation on MET, which has an inhibitory effect on MET signaling [150]. Numerous microRNAs (miRNAs), which are small noncoding RNAs that function by inducing mRNA degradation or translational repression, have also been identified to negatively regulate MET protein expression and impair MET-mediated invasive growth of tumor. These miRNAs include miR-133b, miR-199a*, miR-34b, miR-34c, miR-23b, and miR-198 [6, 151–155].
5.4 The Role of the HGF/SF-MET Axis in HNSCC Progression and Metastasis
HGF/SF-MET signaling alters various cellular responses including mitogenesis, cell motility, and morphogenesis in both embryonic and adult life. Thus, interaction with other cell surface receptors such as EGFR, altered regulation, expression, or over-activation of HGF/SF and/or MET and associated signaling components drives the malignant progression of several different types of tumors including breast cancer, renal carcinomas, colon cancer, and head and neck cancer [17]. In this section, current understanding of the HGF/SF-MET axis in head and neck cancer biology and advances in therapeutic inhibitors targeting HGF/SF-MET signaling will be discussed (refer to Fig. 5.3).
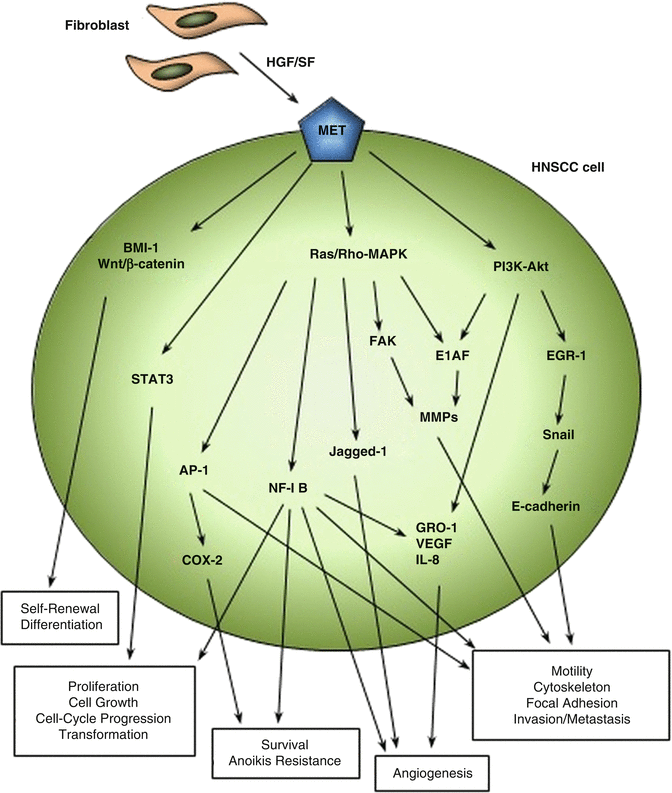
Fig. 5.3
HGF/SF-MET pathways in HNSCC. Major signaling pathways involved in HNSCC progression, invasion, and metastasis due to altered activation or expression of HGF/SF and MET are shown. Tumor-associated fibroblasts are the known major source of HGF/SF in the HNSCC tumor environment. The aberrant expression of MET in HNSCC cells and tissues is highly related to increased frequency of central lymph node metastasis in in vivo experiments and HNSCC patients
Ligand-Dependent Activation of HGF/SF and MET Signaling
MET is frequently overexpressed in nearly all types of HNSCC, correlating positively with more advanced stages of tumor and shortened patient survival. Seiwert et al. reported that immunochemistry of HNSCC tissue and normal adjacent mucosa showed strong MET overexpression in 84 % of the HNSCC tissue samples compared to 21 % of low MET expression in normal tissue [156]. The presence of phosphorylated MET expression also closely correlates with overall expression of MET [156]. Immunoblotting also revealed that 18 of 20 HNSCC cell lines (90 %) express MET [156]. MET inhibition using small-molecule MET tyrosine kinase inhibitors, SU11274 and PF-2341066, downregulates MAPK and PI3K-AKT pathways and results in decreased cell viability, proliferation, and HGF-induced motility in vitro [157, 158]. PF-2341066 inhibits HNSCC tumor growth in vivo with decreased proliferation and increased apoptosis within the tumors [158]. HGF/SF overexpression is also detected in 45 % of HNSCC tumors in mice [157], and the mean serum HGF/SF concentration has been found to be significantly elevated in HNSCC patients [159]. Further, Kim et al. demonstrated that the serum HGF/SF levels are higher in advanced tumor stages and patients with recurrent HNSCC retain higher levels of serum HGF/SF compared to the level at 1 month after the resection of tumor [159]. HGF/SF is secreted by HNSCC tumor-derived fibroblasts [158]. Interestingly, amplification of the MET gene is only detected in 13 % of HNSCC tumor tissues despite the significant overexpression of the MET receptor, suggesting other levels of MET receptor regulation and that HGF/SF predominantly functions in a paracrine fashion to activate MET signaling in HNSCC [158, 160].
Ligand-Independent Activation of HGF/SF-MET Signaling
Several mutations in the MET receptor have been identified in HNSCC: T230M and E168D mutations in the ligand-binding domain, R988C and T10101 mutations in the juxtamembrane domain, and T1275I and V14333I mutations in the tyrosine kinase domain. However, the relative contributions of these mutations to HNSCC progression remain unknown [160]. Two activating somatic mutations in the MET oncogene, Y1230C and Y1235D, are associated with sustained MET activation in HNSCC metastasis [161]. The precise mechanism of MET activation by these two mutations is still unknown; however, it has been proposed that the Y1235D mutation may constitutively activate MET signaling by mimicking phosphorylated tyrosine by substituting tyrosine with the negatively charged aspartic acid. Interestingly, Y1235 is one of the two tyrosines (Y1234 and Y1235) previously identified as a major autophosphorylation site on the kinase domain of the MET receptor [16]. Activating mutations of MET are also found in hereditary papillary renal carcinomas and sporadic renal cancers [162]. Findings by Di Renzo M et al. indicate that HNSCC cells expressing mutant MET have growth advantages and are clonally selected for tumor dissemination and that this aberrant MET expression is highly associated with HNSCC progression and metastasis, as the frequency of mutant MET increases from 2 % in the primary tumors to 50 % in the late-stage metastatic HNSCC [161]. Since HGF/SF expression is highly elevated in HNSCC patients [157, 159], ligand availability is probably not a limiting step in ligand-independent activation of aberrant MET.
HGF/SF and MET Signaling in HNSCC Metastasis
The process of cancer metastasis is an elaborate, yet inefficient one requiring many sequential steps in order to establish secondary tumors at the distant site. Individual tumor cells must first disengage from the primary tumor, migrate through the extracellular matrix, and intravasate into the blood or lymphatic vessels where they can travel to other sites of the body. Before colonizing a tissue, they must then extravasate and utilize enzymes pertinent to the invasive process before they can establish a metastatic tumor [163]. Cervical lymph node metastasis, which occurs frequently in head and neck cancer, serves as an important indicator of local recurrence or distant metastasis of HNSCC [6]. Thus, great effort has been made to understand molecular profiles that may predict HNSCC lymph node metastasis in hopes to develop treatment options that will be effective for patients. The HGF/SF-MET axis stimulates all the signaling pathways that are known to convey proliferative, anti-apoptotic, and pro-migratory phenotypes in development and adult life. These same signaling pathways are activated by HGF/SF-MET in HNSCC tumor progression. As previously discussed, activation of multiple pathways, including MAPK, Ras, and PI3K-AKT, promotes the execution of MET-dependent invasive growth and metastatic phenotypes in cancer cells [16]. Indeed, several studies indicate that MET and/or its ligand HGF/SF overexpression correlates highly with lymph node metastasis, pathologic stage, disease recurrence, and survival of HNSCC patients [5, 7, 164–169]. Anatomically distinct expression patterns of MET have been found in different HNSCC tumors. Hypopharyngeal SCC (HSCC) is associated with the highest MET expression, followed by OSCC, laryngeal SCC (LSCC), and nasal cavity SCC (NSCC) [164]. This finding also correlates with the frequency of lymph node metastasis occurring in HSCC, OSCC, LSCC, and NSCC [164]. Increased MET expression has also been found in pregnant HNSCC patients with invasive tumors compared to pregnant patients with minimally invasive tumors [170].
Nude mice injected with HNSCC cells into the lateral tongue form tumors within 14 days post-injection compared to mice injected with saline or immortalized oral keratinocytes (OKF-TERT1), which failed to form tumors even 6 months post-injection [157]. Moreover, nude mice injected with MET knockdown HNSCC cells had prolonged survival rates compared to the control mice injected with HNSCC cells expressing MET [157]. Elevated levels of HGF/SF have also been positively linked to lymph node metastasis of HNSCC in vivo [7, 166]. Consistent with these in vivo findings, fibroblast-derived HGF/SF stimulates mitogenesis and invasive growth of MET-expressing HNSCC cell lines via induction of tyrosine phosphorylation of FAK (p125FAK), which co-localizes with several components of focal adhesions [171]. FAK is frequently overexpressed in HNSCC [172], and phosphorylation of FAK allows binding of SRC, GRB2, and PI3K and activation of downstream pathways in HNSCC [173, 174]. In addition, transfection of HNSCC cells with a small interfering RNA (siRNA) targeting SRC inhibits MET-mediated activation of ERK1/2-MAPK, STAT3, and PI3K pathways, decreases invasion, and induces apoptosis, suggesting an important role for MET-FAK-SRC signaling in the invasive phenotype of HNSCC cells [175]. Molecular profiling of transformed and metastatic murine SCC cells also reveals altered expression of MET and increased activation of the NF-κB signaling pathway, a downstream signal of HGF/SF-MET axis [176].
Epithelial cells require intracellular adhesion contacts with neighboring cells in order to form continuity, and organization of cells within tissues and the cadherin superfamily, which includes cadherins (found in adheren junctions) and desmocollin and desmogleins (found in desmosomal junctions), are critical for this to occur [177]. Normal keratinocytes typically express E-cadherin and P-cadherin [178]. The role of E-cadherin has been extensively investigated in the breast, colon, bladder, pancreas, prostate, liver, and SCC. Loss of E-cadherin function is associated with tumor invasion and increased mobility of cancer cells in the primary tumor, which is the first step in the tumor invasion process [179, 180]. Transfection of E-cadherin into invasive cells can reverse this invasive phenotype and impair tumorigenesis [181]. Decreased expression of E-cadherin and P-cadherin is significantly correlated with invasive behavior of HNSCC and predicts nodal metastasis [182, 183]. Downregulation of E-cadherin in tumors can be due to hypermethylation of CpG islands within its promoter [184], deletion of the gene located on chromosome 16 [185], and/or upregulation of the transcription factor Snail, which suppresses E-cadherin expression [186]. In HNSCC cell lines, HGF/SF stimulation of MET results in increased expression of Snail through activation of the MAPK pathway and early growth response factor-1 (Egr-1) activity, thereby suppressing E-cadherin expression and translocation from the cell membrane to the cytoplasm [187, 188]. The Snail family of proteins has been found to contribute to epithelial-mesenchymal transition (EMT) in HNSCC by upregulating the expression of ECM-degrading enzymes such as matrix metalloproteinase-2 (MMP-2) and promoting cell invasion [189]. A reverse correlation of E-cadherin and Snail expression has also been reported in HNSCC [190].
Changes in ECM patterns and destruction of the basement membrane, likely due to increased expression and activation of matrix-digesting enzymes, are a feature defining the transition from in situ carcinoma to preinvasive or invasive carcinoma [191]. The basement membrane poses a significant barrier to the movement of tumor cells; thus, an important event in the metastatic process is the degradation of basement membrane components by matrix metalloproteases (MMPs) [192, 193]. MMPs are important in many normal biological processes including embryonic development, angiogenesis, and wound healing, as well as in pathological processes such as inflammation, tissue destruction, and tumor metastasis [192]. MMP activity was first considered to be limited to the invasion and metastasis steps in tumor progression; however, recent studies reveal that MMPs are involved throughout many steps of cancer development and malignancy, regulating apoptosis, promoting cell proliferation and angiogenesis, and escaping immune surveillance [192, 193]. The MMP superfamily has approximately 25 members, and these zinc-dependent endopeptidases can degrade most of the ECM. Most MMPs are secreted out of the cell and diffuse through the ECM; however, a second class of MMPs, the membrane type MMPs (MT-MMPs), are anchored to the cell membrane [192]. Unlike classical oncogenes, upregulation of MMP expression in tumors is mostly due to transcriptional changes rather than genetic alterations [192]. In human HNSCC, cancer cells are not the only source of MMPs. MMP-2, MMP-9, and MT1-MMP are frequently identified in the stromal tissues within HNSCC [194, 195], and fibroblast-derived MT1-MMP can promote tumor progression in vitro and in vivo [196]. Activation of MET by HGF/SF in HNSCC cells leads to recruitment of Rho GTPase and the phosphorylation of FAK (P125FAK), which promotes expression of MMP-2 and facilitates invasive phenotype through mechanisms described in earlier sections of this chapter [79, 171, 173, 174]. In addition, HGF/SF-MET-induced MAPK-ERK1/2 and PI3K-Akt stimulates expression of the Ets-oncogene family transcription factor E1AF to upregulate MMP-1, MMP-3, and MMP-9 [197, 198]. E1AF-mediated transcriptional upregulation of MMP-9 requires binding of chloramphenicol acetyltransferase (CAT) to the Ets-binding site on the MMP-9 promoter [198]. HNSCC cells with a mutant MMP-9 promoter that lacks the Ets-binding site lose almost two-thirds of wild-type CAT activity and are less invasive [198]. MMP expression profiles for HNSCC patient specimens from the Cancer Genomic Atlas show that expression of multiple MMPs including MMP-2 and MMP-9 is elevated in HNSCC patients compared to control tissues [199].
Anoikis is a term for apoptosis induced by inadequate or inappropriate detachment of anchorage-dependent cells from the surrounding ECM [200]. Disruption of normal cell-matrix interactions can induce anoikis to prevent improper migration of cells during normal development and organ regeneration or repair; however, metastatic tumor cells have shown to retain anoikis resistance during EMT and invasive growth [195, 201]. Zeng Q et al. have shown that HNSCC cells undergo anoikis upon loss of matrix contact, but in the presence of HGF/SF, suspension-induced anoikis of HNSCC cells becomes suppressed. This HGF-induced anoikis resistance has been found to be dependent on the activity of AP-1, which is induced by HGF/SF-mediated ERK and PI3K-AKT signaling and is independent of NF-κB activation [202, 203]. To protect HNSCC cells from anoikis, an anti-apoptotic gene, cyclooxygenase-2 (COX-2), is induced by HGF/SF in an AP-1-dependent fashion [203]. Inhibition of COX-2 activity partially prevents HGF-mediated HNSCC cell survival, while overexpression of COX-2 provides resistance against anoikis in vitro and in vivo [203].
AP-1-dependent invasion requires the activation of the ERK1/2 MAPK pathway or the JNK and p38 MAPK pathways induced by HGF/SF-MET signaling in various cancers including HNSCC [74, 75, 203]. In vitro experiments on the human SCC23 head and neck cancer cell line reveal that lysine-specific demethylase 4A (KDM4A, also known as JMJD2A) is a key epigenetic regulator of AP-1 activation induced by HGF/SF-MET signaling [75]. KDM4A aids in HGF/SF-stimulated transcriptional upregulation of JUN and FOSL1 by removing trimethyl lysine 9 of histone H3 (H3K9me3) from their promoter regions, which allows recruitment of AP-1 to the JUN and FOSL1 promoters [75]. This initiates a positive feedback loop that increases expression of c-JUN and c-FOS proteins that dimerize to form more AP-1 [75]. KDM4A-dependent induction of AP-1 also allows for the transcriptional regulation and expression of genes that promote invasive growth and metastasis in HNSCC cells [75]. Deletion of KDM4A significantly inhibits cervical lymph node metastasis in an orthotopic nude mouse model of HNSCC [75]. Moreover, the abundant expression of KDM4A correlates with increased expression of JUN and FOSL1 in human SCC tissues, and KDM4A expression is significantly higher in metastatic lymph node tissues compared to primary SCC tissues, indicating the critical role of KDM4A in activating AP-1 and promoting metastasis [75]. Taken together, compelling evidence indicates the importance of the HGF/SF-MET axis in the metastasis of HNSCC.
HGF/SF and MET in HNSCC Angiogenesis
Inducing angiogenesis is one of the classic hallmarks of cancer [204]. The process of angiogenesis is a normal occurrence that is necessary not only during embryonic development but also in the adult during pregnancy, the female menstrual cycles, and the wound-healing repair processes [205]. Angiogenesis is a highly ordered and tightly regulated multistep process that is dependent on a balance of pro-angiogenic and anti-angiogenic factors [204]. Endothelial cells have a low turnover rate and remain typically quiescent in the adult with a few exceptions aforementioned [204]. However, during tumor progression, angiogenesis is almost always induced and sustained to continually sprout new vessels that help supply oxygen and nutrients for the successful growth and spread of malignant cells [204, 205]. These vessels are often abnormally shaped and leaky, providing easy opportunities for tumor cells to enter the circulation and travel to distant organs or lymph nodes [204]. Tumor angiogenesis is stimulated in tumor cells by a variety of factors secreted into the microenvironment by malignant cells, stromal fibroblasts, and immune cells [205, 206]. The complex interplay between these cell products results in the production of angiogenic factors such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), prostaglandin E2 (PGE2), TGFβ, interleukin-8 (IL-8), angiopoietin-1, and angiopoietin-2 by HNSCC cells [206]. HNSCC-associated fibroblast-secreted HGF/SF or elevated levels of serum HGF/SF increase growth-regulated oncogene-1 (GRO-1), IL-8, and VEGF production from HNSCC cells [207, 208]. Higher serum HGF/SF levels correlate with higher levels of IL-8 and VEGF in patients with HNSCC and an increase in phosphorylation of MET in HNSCC cell lines [209]. HGF/SF-induced MET activation results in phosphorylation of the MEK-MAPK pathway substrate p42/p44erk and PI3K pathway substrate AKT [209]. VEGF recruits endothelial cells from surrounding tissues and stimulates their proliferation in order to form new blood vessels [205]. VEGF and IL-8 are often co-expressed in aggressive HNSCC tumors and are correlated with decreased survival of patients [210]. HGF/SF-MET-mediated IL-8 and VEGF production can be further enhanced upon combinatorial overexpression of EGFR and p53R175H [207], resulting in increased activity of the NF-κB and AP-1 transcription factors [211, 212]. GRO-1 is an autocrine growth factor and a member of the C-X-C chemokine family that is upregulated by NF-κB and promotes angiogenesis, tumorigenesis, and metastasis in HNSCC in vitro and in vivo [94, 176, 208].
Neurogenic locus notch homology protein 1 (Notch 1) is a transmembrane receptor that is involved in numerous biological functions, including vascular formation during early embryogenesis [213, 214]. In humans, there are four membrane-bound receptors (Notch 1-4) and two families of ligands, Delta like (Delta-like 1, 3, and 4) and Jagged (Jagged-1 and Jagged-2) [213]. Notch signaling is activated through contact-dependent interaction between cells expressing Notch receptors and adjacent cells expressing Notch ligands at the surface [214]. Recent studies demonstrate that HGF/SF initiates cross talk between HNSCC cells and neighboring endothelial cells that express the Notch 1 receptor. HGF/SF-MET activates the MAPK signaling in HNSCC cells which in turn results in upregulation of Jagged-1 expression on the cell surface. This binding of Notch 1 on the endothelial cells and Jagged-1 on HNSCC cells promotes endothelial cell tube formation in vitro and tumor angiogenesis and tumor growth in a xenograft nude mouse model [214].
Although HNSCC tumors tend to be vascularized, they also have enhanced lymphatic draining. This allows neoplastic cells to take advantage of these characteristics to spread locally and regionally [215]. Lymphatic spread is the primary mechanism for HNSCC metastasis, and patient mortality is generally due to locoregional spread rather than distant metastasis [215]. Patients with extracapsular nodal spread of tumor cells have a poorer prognosis and are three times more likely to die from regional recurrence within 5 years [216]. In recent years, much attention has been given to lymphangiogenesis as a mechanism to promote dissemination of tumor cells. Similar to hemangiogenesis, lymphangiogenesis entails the growth of lymphatic vessels from preexisting vessels by recruitment of lymphatic endothelial cells (LECs) [217]. A strong correlation exists between lymphatic microvessel density and lymph node metastasis [218]. Lymphatic capillaries are composed of a single layer of endothelial cells, with no surrounding smooth muscle, pericytes, or a regular basement membrane [218]. They are anchored to the ECM by fibrillin filaments that cause dilation, rather than collapse, upon changes in hydrostatic pressure [217, 218]. This unique composition might allow tumor cells to enter and exit the lymphatic system even more efficiently. The process of lymphangiogenesis is governed by molecules secreted by tumor cells or stromal cells within the local environment such as VEGF, neuropilin-2, angiopoietin, basic fibroblast growth factor (bFGF), and insulin-like growth factor (IGF) [219, 220]. VEGF isoforms C and D are predominantly involved in LEC proliferation and migration, while hemangiogenesis mainly involves VEGF-A [221]. HGF/SF can promote lymphatic tube formation by influencing lymphatic endothelial cell proliferation and migration [222]. Thus, further investigation on the role of HGF/SF-MET-mediated lymphangiogenesis is urgent to fully understand the mechanisms of HNSCC metastasis and develop effective therapeutic modalities.
HGF/SF and MET Signaling in Cancer Stem Cells
Human HNSCC cells overexpressing MET (Met+ HNSCC cells) have a capacity to self-renew, to form spherical colonies in anchorage-independent culture conditions, and to generate tumors that recapitulate the heterogeneity of the parental tumors, exhibiting stem-like properties in vitro and in vivo [223–226]. These cells are also cisplatin resistant and promote in vivo metastasis [223]. Isolated Met+ HNSCC cells using a second marker, CD44, further enhances the in vivo tumorigenicity [223]. In addition, Met+ HNSCC cells upregulate expression of the developmental gene, polycomb ring finger oncogene (BMI1), which has been shown to be a critical player in the maintenance and self-renewal of both normal stem cells and cancer stem cells [227, 228] and in the invasive potential of epithelial malignancies, including HNSCC [223]. Small hairpin RNA (shRNA)-mediated knockdown of MET in Met+ HNSCC cells inhibits stem-like properties and downregulates Wnt/β-catenin signaling, while pharmacologic inhibition of MET with PF-2341066 and the concurrent use of the conventional chemotherapeutic agent, docetaxel, synergistically improves the efficacy of docetaxel and reduces metastasis in a mouse xenograft model of HNSCC [226]. Consistent with these findings, Wnt/β-catenin signaling is also highly active in stem-like glioblastoma cells enriched with MET expression, and Wnt/β-catenin signaling is directly modulated by HGF/SF-dependent MET activation [229]. Thus, understanding how MET interacts with and activates the Wnt/β-catenin pathway is critical to determine how these molecules confer stem-like properties in HNSCC such that they may be considered as potential therapeutic targets.
HGF/SF-MET-Mediated Resistance to HNSCC Therapy
Patients with high MET expression tend to a have poor response to both radiotherapy and chemotherapy [229, 230], and low expression of MET has become a predictive factor for a positive response to chemotherapy in HNSCC patients [230]. HGF/SF-MET signaling has been shown to interfere with ionizing-radiation-induced apoptosis by downregulating B-cell lymphoma-extra large (BCL-xL), an anti-apoptotic member of the B-cell lymphoma 2 (BCL-2) family, and enhancing DNA repair through the activation of the PI3K-Akt signaling pathway in epithelial carcinoma cells of the breast and prostate [231]. A cohort study evaluating 97 patients with radically irradiated HNSCC also shows immunohistochemical expression of MET and BCL-xL, and this is correlated with decreased rates of complete remission of the primary tumor [229]. Moreover, the same study shows that high MET expression in patients is associated with low disease-free survival and low overall survival [229], suggesting that MET and BCL-xL are attractive targets for radiosensitization in HNSCC.
Numerous studies report that the MET signaling pathway may be involved in acquired resistance to anti-EGFR therapy through cross talk with the EGFR signaling pathway [6, 19, 20]. EGFR is one of the most widely overexpressed tyrosine kinase receptors in HNSCC tumors (over 80 %) and has been identified as a poor prognostic factor for HNSCC patients [8]. Many anti-EGFR small-molecule inhibitors or monoclonal antibodies against HNSCC have been developed; however, EGFR-targeted therapy remains modestly effective in treating HNSCC due to acquirement of resistance through unidentified mechanisms [232]. Recently, the MET signaling pathway has been recognized as a contributing factor to resistance to EGFR inhibitors. Over-activation of MET correlates with resistance to gefitinib, a selective potent EGFR inhibitor, in lung cancer [233]. Similarly, MET activity increases with the use of the EGFR-neutralizing antibody cetuximab or other EGFR inhibitors in HNSCC cell lines [6]. The MET receptor and EGFR share many signal transduction components and downstream pathways, and because of this cross talk, it has been reported that MET may substitute for and bypass EGFR signaling in HNSCC when EGFR is inhibited [234]. For example, aberrant expression of cortactin, a key regulator of dynamic actin networks and modulator of receptor signaling, can promote MET activation and is associated with enhanced HNSCC cell proliferation and resistance to the gefitinib [235]. Furthermore, dominant-active c-src in HNSCC cells results in constitutive activation of MET and confers resistance to erlotinib, an EGFR tyrosine kinase inhibitor [236].
5.5 Therapeutic Strategies Against HGF/SF-MET Signaling in HNSCC
Due to the broad biological impact of HGF/SF-MET signaling in HNSCC cancer pathogenesis and progression, extensive efforts have been made to develop agents against HGF/SF and MET (summarized in Table 5.1). These include receptor antagonists and decoy molecules, small-molecule tyrosine kinase inhibitors (TKIs), or humanized monoclonal antibodies (mAb).
Table 5.1
Current therapeutic strategies targeting HGF-MET signaling in HNSCC
|
Name of the drug
|
Class of the drug
|
Primary targeting molecule
|
Stages of development
|
References
|
|---|---|---|---|---|
|
NK4
|
Biological antagonist
Truncated isoform of full-length HGF/SF
|
MET
|
Preclinical
|
|
|
Tivantinib (ARQ179)
|
Selective small-molecule MET tyrosine kinase inhibitor
|
MET
|
Phase I/II
|
|
|
Foretinib (XL880)
|
Broad-spectrum small-molecule tyrosine kinase inhibitor
|
MET, VEGF
|
Phase II
|
|
|
LY2801653
|
Broad-spectrum small-molecule tyrosine kinase inhibitor
|
MET, MST1R
|
Phase I
|
[253]
|
|
Cabozantinib (XL184)
|
Broad-spectrum small-molecule tyrosine kinase inhibitor
|
MET, VEGFR2, RET
|
Phase III
|
[254]
|
|
Crizotinib (PF-02341066)
|
Selective small-molecule MET tyrosine kinase inhibitor
|
MET, ALK
|
Phase III
|
|
|
Rilotumumab (AMG102)
|
Humanized monoclonal antibody
|
HGF/SF
|
Phase I/II
|
|
|
Onartuzumab (OA5D5 or MetMAb)
|
Humanized monovalent antibody
|
MET
|
Phase III
|
|
|
SAIT301
|
Humanized monoclonal antibody
|
MET
|
Preclinical
|
[262]
|
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


