Fig. 11.1
Normal coagulation. (a) Without injury; (b) Vessel injury with activation of vWF and recruitment and activation of platelets followed by; (c) activation of the clotting cascade on the platelet surface; and (d) fibrin clot formation
This complex network of factors, working in concert on a continual basis in vivo, is a necessity for the survival of the host. Cancers can utilize this intricate pathway in such a fashion as to promote tumor growth and metastasis (Fig. 11.2). Excessive activation of this mechanism leads to pathological clotting and occlusion of vessels. Increased vessel occlusion, which is characteristically increased in patients with cancer, suggests that cancer is a prothrombotic state. This prothrombotic state may provide a microenvironmental niche, which promotes tumor growth and metastasis.
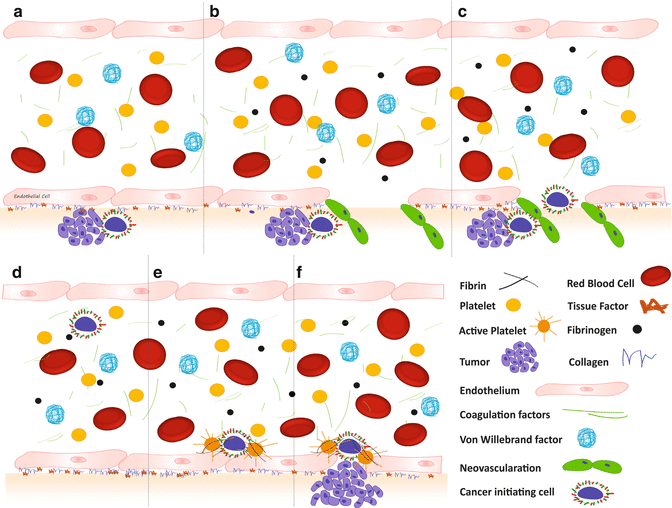
Fig. 11.2
Initiation of metastatic disease. (a) Subendothelial tumor. (b) Induction of angiogenesis, including (c) vascular invasion; (d) hematogenous spread; (e) adhesion; and (f) invasion
11.4 Evidence Suggesting Coagulation System Is Active in the Tumor Microenvironment and Involved with Metastasis
A majority of cancer patients have abnormalities in their coagulation systems, predominantly characterized by a hypercoagulable state [14]. Such findings are supported by several studies which have shown a number of procoagulation factors to be significantly elevated in patients with cancer [15–17]. In addition, cancer cells are capable of modulating the hemostatic system by virtue of intrinsic prothrombotic, fibrinolytic, and platelet-aggregating properties. They can stimulate the host inflammatory responses as a result of tumor-host interaction that can further impact the coagulation cascade. The evidence supporting the role of key coagulation factors in tumor growth and metastasis is elaborated below.
11.4.1 Fibrin
Fibrin is the classic end product of coagulation and was discovered to be an important matrix for tumor cell growth. Its precursor, fibrinogen, is first cleaved by the serine protease thrombin (particularly at the site of vascular injury). An additional function of thrombin is the activation of FXIII, which is then able to convert soluble fibrin into an insoluble form. Both fibrinogen and fibrin have been noted to be elevated in the tumor stroma, indicating extravascular activation of the hemostatic process, which is favored (in part) by the increased vascular permeability of newly formed vessels [18]. The fibrin gel provides a provisional matrix that can sequester angiogenesis-stimulating growth factors that promote cancer cell expansion and metastasis. This complex matrix also provides a surface for prothrombinase assembly by platelets inside the blood vessel. In this setting, proangiogenic growth factors including vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and insulin-like growth factor (IGF) may be protected from degradation within the fibrin matrix [19]. Intriguingly, tumor cells are capable of controlling their own microenvironment by constitutively synthesizing and secreting fibrinogen [20]. In addition to the abovementioned mechanisms, it has been demonstrated that fibrin can induce tumor cells derived from lesions of the oral cavity to express the proangiogenic interleukin IL-8 in vitro [21].
The fibrin mesh can also contribute to metastasis. Aggregates of tumor cells and platelets are rich in fibrin and can further promote platelet adhesion to tumor cells and facilitate metastatic dissemination [22]. From a clinical perspective, it is noteworthy that patients with gall bladder carcinoma have been shown to be hyperfibrinoginemic and have poor prognoses [23]. Studying the interaction of fibrin with the tumor cells will enhance our understanding of the tumor microenvironment.
11.4.2 Factor V
Factor V, which circulates in an inactive form in the bloodstream, is primarily made in the liver. When factor V is activated, along with factor X, prothrombin is converted to thrombin. Bruggemann et al. have shown that mice with factor V Leiden, and a consequent prothrombotic phenotype, are prone to the development of metastases [24]; however, this has not been consistently replicated in human studies. When comparing the prevalence of the factor V Leiden gene mutation in patients with gastric cancer to the general population, significant increases were not identified [25]. Similar results were obtained when oral cancer patients were compared to healthy controls [26]. Patients with colon cancer, however, demonstrated increased prevalence of factor V Leiden (27 %) in 44 patients with colorectal cancer compared to 6 % in 600 healthy individuals [27]. Furthermore, the risk of colon cancer decreased by 30 % in heterozygous factor V Leiden patients compared with non-carriers. This might be explained by a level-dependent effect of thrombin on carcinogenesis via protease-activated receptors (PARs) as both PAR1 and PAR4 are associated with colon carcinogenesis. Activation of PAR4 requires a higher level of thrombin than PAR1 activation and is more sustained than PAR4. Also low levels of thrombin alone can induce endothelial barrier protection suggesting that PAR1 activation on endothelial cells has opposite biological effects [27]. Larger population-based studies are needed to shed more light on this apparent incongruity.
11.4.3 Thrombin
Thrombin is central to the coagulation pathway, with the primary endpoint being the formation of the fibrin clot. In the normal physiological state, serum concentrations of thrombin are kept in check (low) largely by a stoichiometric balance with its natural inhibitor, antithrombin III. During coagulation, the prothrombinase complex converts prothrombin to thrombin to increase levels that overcome antithrombin III. Interestingly, the presence of a malignancy has the capacity to increase endogenous thrombin, and it has been shown that tumor cells that constitutively express tissue factor can combine with factor VIIa to promote the generation of thrombin. An additional method by which thrombin has been shown to promote metastasis is through its ability to increase platelet-tumor adhesions. Thrombin has also been shown to increase lung metastasis in experimental mouse models [28]. Recently it has been shown that thrombin activates PAR-1 on the stromal cells adjacent to colon cancer cells thereby enhancing tumor growth.
11.4.4 Tissue Factor/Factor VII
Tissue factor is expressed by tumor cells and interacts with coagulation factor VII and factor X and generates thrombin (extrinsic pathway), thus enhancing tumor progression. As outlined above, thrombin is a driving force behind tumor growth and metastasis. More recently, Koga et al. have taken advantage of the fact that pancreatic cancer cells express tissue factor in the development of an antibody drug conjugate (ADC) wherein the drug is conjugated to an anti-tissue factor antibody. Treatment with this ADC led to internalization and targeted action against cancer cells both in vitro and in a mouse model [29].
11.4.5 Factor VIII
Several clinical trials have produced conflicting results with the use of anticoagulants in cancer outcomes; Langer et al. and Bruggemann et al., however, have published studies demonstrating that congenital coagulation disorders dramatically influence the ability of cancers to grow and metastasize. Langer and colleagues injected a murine model of hemophilia A with B16F10 melanoma cells. While these cells produced metastases in the lungs, the metastatic potential was significantly enhanced when human FVIII was administered. This study also revealed that administration of lepirudin (a direct thrombin inhibitor) significantly reduced metastasis [30]. Bruggemann and his colleagues demonstrated that the metastasis of murine melanoma cells was diminished in FVIII knockout mice compared to their wild-type littermates and that factor V Leiden mice had more lung metastases than that their littermate controls. Recent literature suggests that coagulation factor deficiency might inhibit cancer metastasis through decreased activation of thrombin. A review of epidemiological studies reporting the causes of death in 8000 hemophilia patients (including 2700 HIV-negative patients) reported a virus-independent cancer rate of 8–16 %. Analysis of the corresponding standardized mortality ratio (SMR) supports the hypothesis that cancer rates, unaffected through HIV or virus-associated liver disease, are decreased in hemophilia patients when compared with the general population [31]. However, other studies have not been able to confirm this trend. Most recently Huang et al. published a review of the incidence and survival rates of cancers among 1054 hemophilia patients. This study showed an increased incidence of cancer in patients with hemophilia [32]. Since most of these studies are retrospective, it is possible that they over- or underestimate the SMR.
11.4.6 Factor XIII
Factor XIII is a thrombin-activated plasma transglutaminase zymogen. It circulates as a heterotetramer composed of two catalytic A subunits and two non-catalytic B subunits. It is best known for stabilizing the platelet-fibrin thrombus. Palumbo et al. compared tumor growth and metastatic potential between wild-type mice and those lacking the active FXIII-A subunit. The loss of function of this subunit significantly decreased the hematogenous metastatic potential in immunocompetent mice. The presence of factor XIII-A impedes natural killer (NK) cell-mediated clearance of tumor cells [33].
11.4.7 Urokinase-Type Plasminogen Activator/Plasminogen Activator Inhibitor-1
The plasminogen-plasmin (P-P) system has an interesting role in tumor metastasis. Urokinase-type plasminogen activator (uPA) has been localized to the “invading front” of both primary and metastatic tumors suggesting uPA might promote tumor migration and invasion by the conversion of plasminogen to plasmin. Plasmin activates pro-metalloproteinases to metalloproteinases that degrade the extracellular matrix and promote invasion. Although it was hypothesized that plasminogen activator inhibitor-1 (PAI-1) would inhibit metastasis by inhibiting the action of uPA, the contrary was observed. Studies have also demonstrated that PAI-1 inhibits apoptosis, increases tumor cell adhesion, and promotes tumor cell proliferation and angiogenesis [34].
11.4.8 Protein C
The role of protein C is directly related to the hypothesis that a prothrombotic state favors metastasis. Overexpression of endothelial protein C receptor (EPCR) in mice resulted in markedly reduced metastases to the lung and liver [35]. This observation was attributed to the interaction and activation of EPCR by aPC and the subsequently decreased expression of endothelial adhesion molecules. EPCR-dependent PAR1 activation by aPC on breast cancer cells promoted cell migration and prevented apoptosis of lung cancer cells [36].
11.4.9 Von Willebrand Factor
Perhaps one of the most intriguing clotting factors known to play a crucial role in tumor metastasis is von Willebrand Factor (vWF). vWF is a prominent plasma protein involved in blood coagulation. Mochizuki et al. found that ADAM28 binds to and cleaves vWF and demonstrated that physiological levels of vWF induced apoptosis in cancer cells. In vitro experiments demonstrated that a rapidly growing metastatic cell line that expressed high levels of ADAM28 was resistant to vWF-induced apoptosis; however, apoptosis occurred when ADAM28 expression or activity was blocked [37–41]. This study also demonstrated that vWF induced cancer cell apoptosis via the alpha V beta 3 (αVβ3) integrin pathway, which ultimately led to the activation of mitochondrial-mediated (intrinsic) apoptosis. They additionally found that the reduction of ADAM28 activity in cancer cells resulted in diminished metastasis. The authors concluded that cleavage of proapoptotic vWF by ADAM28 enhanced lung metastasis by promoting cancer cell survival within the circulatory system. It was also noted that ADAM 28 was able to cleave native and denatured vWF (in contrast to ADAMTS13) after intravasated tumor cells interacted with platelets and the vessel wall before extravasation from the circulation. vWF has been implicated in the binding of platelet tumor cell thrombi to the endothelium. Human tumor cells increase endothelial cell production of vWF, thereby enhancing adhesion of cancer cells to the endothelium. Contrary to expectation, cancer cell culture studies revealed vWF did not influence tumor cell proliferation or invasion but instead induced apoptosis. Consistent with the above findings, vWF factor-null mice had increased numbers of pulmonary metastases. Following restoration of plasma vWF level by injection, metastasis was inhibited.
Recent developments have shown many biological functions of vWF. A regulatory role for endothelial vWF in angiogenesis has been hypothesized, as vWF is closely related to other well-known regulators of vascularization. In the clinical setting, it has been noted that there is increased incidence of angiodysplasia in patients with von Willebrand disease. Starke et al. used short interfering RNA (siRNA) to inhibit vWF expression in human umbilical vein endothelial cells (HUVEC) and observed increased angiogenesis and improved vascular endothelial growth factor receptor-2 (VEGFR-2)-dependent proliferation and migration. VEGFR-2 is an important receptor for the key angiogenesis mediator hormone VEGF-A. vWF-deficient mice have increased angiogenesis and a larger vascular network. Contrary to the findings mentioned in the above studies, vWF can also function as promoter of tumor cell metastasis. Studies have shown that B16-BL6 melanoma cells adhere to vWF through membrane-expressed integrin αVβ3. Many clinical studies have shown significantly elevated levels of plasma vWF in a variety of cancers. It has been reported that during the process of tumor angiogenesis, platelets produce VEGF which promotes endothelial proliferation and elevated plasma levels of vWF. vWF is essential for the formation of WPBS in endothelial cells which store a number of proteins including Ang-2. It was speculated that the loss of vWF might result in the ability of the endothelial cells to store Ang-2 and hence increase its dysregulated release. Ang-2 is known to promote VEGF-dependent stimulation of EC sprouting and migration and has also been found to induce derogation of integrin β3. Therefore, deregulated Ang-2 release from vWF-deficient ECs could be involved in the proangiogenic effects of vWF. Additionally, increased levels of Ang-2 were found in the supernatant of vWF siRNA-treated cells [37–41].
11.4.10 Platelets
Platelets are important cells in the coagulation pathway, and platelet turnover has been noted to be enhanced in patients with cancer. The function of platelets in coagulation and hemostasis are repeated during tumor metastasis. In the coagulation cascade, platelets are activated by thrombin. Platelets are known to assist in tumor cell arrest at the vessel wall (adhesion) and survival within the vasculature (immune evasion). They also help with extravasation (leaving the vascular system) and tumor cell survival and proliferation at the sites of the metastasis [42]. Platelets have also been shown to release VEGF and promote angiogenesis. It has been noted that both pro- and antiangiogenic molecules are present in the alpha granules which are released based on different stimuli.
The risk of development of thrombosis is very high in pancreatic and head and neck cancer [43]. These cancers are metastatic, extremely aggressive, and resistant to therapy. HNSCC cells secrete PAI-1 that, in addition to promoting thrombus formation, can inhibit apoptosis and promote angiogenesis. Increased secretion of urokinase, which further serves to promote proliferation and invasion through the degradation and reformation of extracellular matrix, is also a feature of these cells [34, 44]. Tissue factor (TF) expression has also been reported in head and neck cancers [45, 46]. TF complexes with activated factor VII to elicit signaling through PAR-2 (a protease-sensitive receptor) that works with platelet-derived growth factor to synergistically drive angiogenesis [47]. Additionally TF can directly increase VEGF signaling while decreasing antiangiogenic thrombospondin signaling [48].
11.4.11 Microparticles
The cells being activated or undergoing apoptosis release procoagulant microparticles (MPs). These may be derived most commonly from platelets, but may also be derived from endothelial cells, erythrocytes, monocytes, lymphocytes, or cancer cells. They are usually defined by their size and range from 0.1 to 1 μm. Surface expression of phosphatidylserine (PS) is also a feature of some MPs and was reported to enhance thrombogenesis 50–100 times compared to the same surface area of platelets. Further clinical and experimental studies demonstrated that TF bearing MPs that shed from tumor cells or monocytes might play an important role in the prothrombotic state associated with malignancies. However, it remains to be seen whether these MPs are directly involved in tumor growth and metastasis [49].
11.4.12 Neutrophil Extracellular Traps (NET)
More than a decade ago, NETs were first described in relation to bacterial killing. Upon activation, neutrophils were noted to release granule proteins and chromatin that together form fibers that bind bacteria. This fibrous structure was necessary for the sequestration and killing of bacteria by delivering a high local concentration of molecules toxic to the bound microbes [50]. This was later found to act as a scaffold for platelet binding and activation, leading to platelet aggregation and thrombus formation [51]. Later, it was shown that in murine models of chronic myelogenous leukemia (CML) and mammary and lung cancer models, there was an increased tendency toward NET formation [52]. The same group later showed that peptidylarginine deiminase 4 (PAD4)-deficient mice, which are incapable of citrullinating histones, demonstrated a lower incidence of DVT compared to wild-type mice [53].
11.5 The Effects of Hemostasis on the Development and Initiation of Cancer
Several disparate lines of evidence support an important role for the coagulation system in the development and propagation of HNSCC [54]. PAI-1 plasma levels are highly dependent on a promoter region polymorphism, with people with the 4G/4G promoter producing more PAI-1 and having a higher incidence of head and neck cancers [55], higher stage disease [56], and a worse prognosis [57]. Tobacco smoke carcinogens increase PAI-1 expression and induce epithelial-mesenchymal transition (EMT) [58]. Thrombin, perhaps the most central factor in hemostasis, has been shown to promote tumor growth metastasis and angiogenesis [28]. Interestingly, several “natural experiments” retrospectively or longitudinally comparing people with hypercoagulable predispositions [59] (e.g., hemophilia) [31] demonstrated a correlation between a procoagulant tendency and increased risk of cancer and cancer mortality. Activated platelets release VEGF and inflammatory mediators at the site of activation potentially contributing to tumor propagation [60], and platelet activation can promote metastasis in tongue cancer [61]. Supporting the importance of this role of platelets in tumor development are recent lines of evidence demonstrating decreased cancer incidence in patients taking the potent antiplatelet agent aspirin for protection against vascular disease and thrombosis, including a benefit of decreased HNSCC [62].
11.6 Novel Assays to Assess the Roles of Coagulation in Cancer Growth and Metastasis
11.6.1 Microfluidics and Global Coagulation Assays in the Study of Hemostasis
Conventional coagulation assays, such as the prothrombin time, activated partial thromboplastin time, or platelet function assays, involve compartmentalized assessments of a single element of the coagulation system or a small number of elements. While these assays provide important information, they make no assessment of critical interactions between various cellular and fluid phase elements of coagulation and no estimation of the contributions of the endothelial compartment and cannot elucidate changes that occur under conditions of flow and sheer stress. All of these interactions become vitally important when examining the role of coagulation in tumor metastasis. Several commercial and “home brew” microfluidic systems have been developed that have similar designs, utilizing air compression and electropneumatic regulators to allow for flow through chambers designed for florescent microscopic and real-time examination. The rate of flow can be manipulated in such systems to modulate the sheer properties over a range of substrates, including endothelial cells. While still at a very early stage, these systems have already provided important insights into the biology of tumor metastasis and the effect of tumor cells on coagulation [63]. Our preliminary studies have indicated that the addition of cancer cells (as a substrate) increased platelet adhesion under a variety of flow conditions. Consistent with the hypothesis that mediators of coagulation might serve as drivers of metastasis, increased adhesion was observed with substrates that utilized human cell lines derived from metastatic tumors compared to cell lines from primary sites (data not shown).
11.6.1.1 Thromboelastography
Thromboelastography (TEG) is a method to examine all the elements of thrombus formation and fibrinolysis in a whole blood samples. A major advantage of TEG, compared to other more traditional compartmentalized blood clotting assays, is that it maintains all the elements (save endothelium) that are present physiologically. In the TEG assay, citrated whole blood samples are recalcified in a chamber with a vibrating probe where tissue factor is then added to induce coagulation. As thrombus forms, the vibration becomes restricted; the degree of this limitation can be quantitated as depicted in Fig. 11.3a. The TEG curve is represented by the following parameters: (i) R (reaction) time (from the start of the test until clot formation) and is expressed in minutes; (ii) α-angle, the angle between the baseline and the tangent to the TEG curve, reflecting the rate of clot formation (thrombin burst); (iii) maximal amplitude (MA) which is a direct measure of the strength of the clot and reflects the total contribution of platelets and fibrin; and (iv) Ly 30, which is the difference between MA and the amplitude after 30 min and reflects the degree of fibrinolysis at 30 min. Dynamic properties of clot formation are assessed by transformation of the TEG profile data into a velocity curve (first derivative of clot strength versus time, V curve), assessing maximum rate of thrombus generation (MRTG), which represents the peak of the first derivative of the velocity of the increase in clot strength, and time to MRTG (TMRTG) which represents initiation of clot formation (lag time) plus the time for clot development to reach maximum rate of clot strength development.
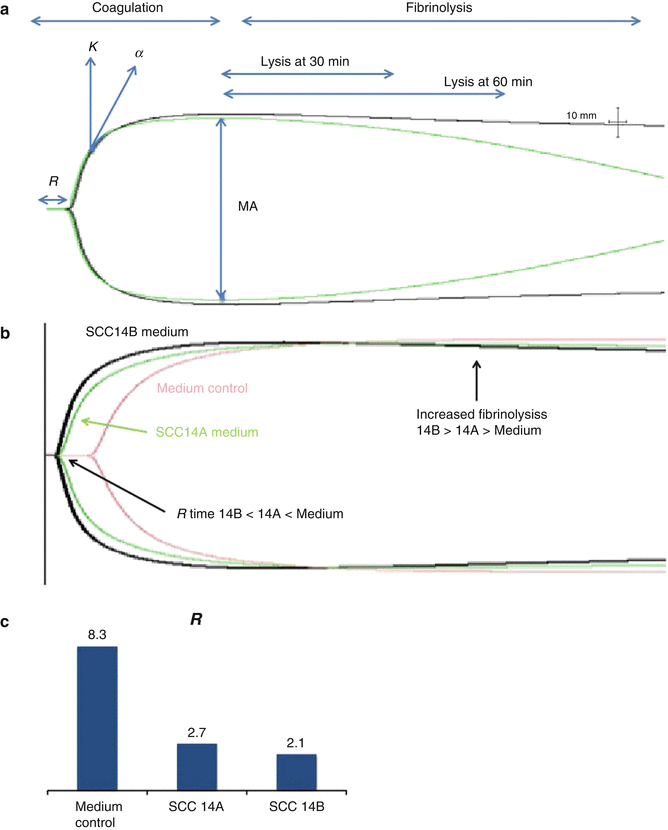
Fig. 11.3
Soluble factors from metastatic HNSCC cells decrease coagulation time. (a) Normal thromboelastography (TEG) tracing demonstrating parameters of analysis for clot lysis at 30 and 60 min: where R reaction time, k coagulation, α angle (speed of clot formation), MA maximum amplitude. (b) TEG demonstrating faster clot formation and fibrinolysis with metastatic UMSCC14B versus primary UMSCC14A cells or medium control. (c) Quantitation of reaction time (R)
We compared paired OSCC cell lines derived from the same patient at diagnosis and relapse/metastasis (UMSCC-10A and UMSCC-10B, UMSCC-14A and UMSCC-14B). Cells were grown to 70 % confluence, and media were replaced with serum-free media for 24 h. When the cells and media were harvested, the medium from each sample was concentrated 5:1, and thromboelastography was performed with control whole blood from a health volunteer and 10,000 cancer cells in 20 μL PBS or 20 μL of concentrated medium plus whole blood. Similar to our observations noted above using sheer stress, the time to clotting (R) decreased with relapsed/metastatic cell lines (Fig. 11.3b, c). These data implicate a procoagulant role for HNSCC cells and stable soluble factors they secreted in culture. Studies are currently underway to identify these factors.
11.6.2 Tumor Spheres
In 1951 George Gey revolutionized the study of cancer when he established the first transformed human cancer cell line (HeLa) for in vitro use [64]. In the decades since, the field of cancer research has relied on nearly identical, two-dimensional (2D) monolayer cell culture models of human tumors that have been primarily generated from biopsy-isolated cells and surgically excised tumors. In fact, nearly 2000 human tumor-derived cells lines exist among repositories worldwide [65]. While the 2D approach has afforded momentous insights into cancer biology, critics have cogently argued that such a “flattened” approach does not sufficiently approximate tumor architecture nor permit the recapitulation of specific physiological niches of the tumor microenvironment required to translate benchtop findings to the clinic. Rather, that cell lines merely reflect the population of cells selected to survive and grow in culture; furthermore, it has been stated that a cell line may simply represent “a snapshot of the tumor at the time the biopsy was taken.” Not only is the tumor heterogeneity lost in immortalized cultures, but also mutations acquired in the course of culture open the door to the possibility of gross over- or underestimation of experimental observations and provide for significant genetic differences between the “same” cell line archived by different laboratories [66, 67].
Concerns for the limitations of studying cancer in monolayers, specifically with regard to the identification of progenitor (stemlike) cells and to the discovery of biomarkers and new anticancer therapeutics, have inspired efforts to develop tumor organ culture and tumorsphere models that more closely simulate tumor anatomy and physiology. Tumor organ culture and sphere formation assays are three-dimensional (3D) culture approaches that take into account the spatial organization of different cell types, permit vascularization, and provide a platform to support intracellular communication and nutrient networks [68]. Sphere-forming assays were first employed by Reynolds and Weiss as an in vitro tool to dissect neuronal networks and isolate stemlike cells [69]. They demonstrated that when neural cells were grown on a low or nonadherent scaffolds at low densities in the absence of serum and in the presence of a precise cocktail of growth factors, cellular spheres would form. These floating spheres were directly correlated to individual stem (progenitor) cell clones [69]. While 2D tissue culture does not readily permit the identification of tumor progenitor cells, enzymatic and mechanical dissociation of tumorspheres has been successfully used to isolate early tumor progenitor cells in many cancers, including HNSCC [70–72].
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


