Fig. 7.1
EGFR signaling pathway and alternative signals. Even with successful EGFR inhibition, substitute signaling pathways can be activated by signaling through other cell-surface receptors including IGF-1R, IL-6R/gp130, c-MET, or VEGFR. Activation of STAT3 by IL-6R/gp130 involves phosphorylation of Janus-activated kinases (JAK), whereas EGFR activates STAT3 directly or through the Ras/Raf/Mek/Erk pathway [46]. STAT3 may be negatively regulated by suppressors of cytokine signaling (SOCS). Constitutive activation of STAT3 plays a causative role in many processes in cancerogenesis through the overexpression of cyclin D1, VEGF, bFGF, HIF-1α, MMP-2, MMP-9, IP-10, GLUT1, Twist, or RANTES
SET protein (I2PP2A or TAF1β) is an inhibitor of PP2A and is responsible for the activation of Akt, c-Myc, and β-catenin pathways [25]. A recent proteomic analysis of HNSCC samples identified that the SET protein is accumulated in oral tumors [26, 27]. SET has multiple proposed functions such as promoting Rac1-induced cell migration [28], chromatin remodeling [29], and sensing of oxidative stress that supports cell survival in HNSCC [27]. On the other hand, stable SET knockdown in HN12 cells stimulated epithelial-mesenchymal transition (EMT) and invasion [30] and caused an increase in the autophagic proteins Beclin-1, LC3B, and p62 [31]. Xi et al. found that the PP2A inhibitor cantharidin activated the unfolded protein response (UPR) and induced apoptosis in a panel of HNSCC cells and other cancers. Although cantharidin may exert its cytotoxic effect through additional mechanisms and the role of the UPR in cancerogenesis is not fully elucidated, it is possible that PP2A inhibition could have contradictory roles at different stages of HNSCC cancerogenesis [32].
STAT3 is another important proliferative and survival signal that is constitutively activated in HNSCC [33–35]. Constitutive activation of STAT3 plays an important role in many processes during cancerogenesis by modulating increased expression of cyclin D1, VEGF, bFGF, HIF-1α, MMP-2, MMP-9, IP-10, and RANTES [33, 36, 37]. HNSCC patients express higher levels of STAT3 in both tumor and histologically normal adjacent tissues compared with epithelium of healthy control subjects. STAT3 protein levels were 9-fold higher in tumor-adjacent mucosa from cancer patients and 11-fold higher in HNSCC tumors compared with healthy controls [35]. STAT3 activation may be mediated by either EGFR or interleukin-6 (IL-6).
IL-6 was increased in the saliva of oral cancer patients (OSCC) [38] and was overexpressed in tumor tissues and sera of patients with HNSCC when compared with the healthy individuals [39]. Multivariate analysis revealed that postsurgery IL-6 concentrations in saliva were a risk factor for locoregional recurrence in patients with OSCC [40, 41]. Salivary IL-6 and TNF-α concentrations were notably higher in individuals with oral leukoplakia in comparison with the healthy subjects [42], and markedly higher levels of IL-8, IL-6, IL-1, and TNF-α were found in OSCC patients’ saliva when compared with subjects with dysplastic oral lesions and healthy controls [43]. Higher IL-6 serum levels were also observed in patients with OSCC compared to healthy controls, results that were confirmed at both the mRNA and the protein levels [44]. Serum IL-6 levels were also markedly higher in patients with higher TNM stage, higher pathological stages, positive bone invasion, and increased tumor depths. Patients with high pretreatment levels of IL-6 (>1.35 pg/mL median level) had worse prognoses for 5-year overall and disease-specific survival [45].
Activation of STAT3 by IL-6R/gp130 involves phosphorylation of Janus-activated kinases (JAK) [19, 20], whereas EGFR activates STAT3 directly or through Ras/Raf/Mek/Erk signaling [46]. Conversely, STATs may be negatively regulated by suppressors of cytokine signaling (SOCS) [47]. Epigenetic silencing of SOCS1 and SOCS3 has been recently reported in HNSCC [48, 49]. SOCS3 expression is reduced early in tumor development, and the overexpression of SOCS3 in HNSCC cell lines diminished proliferation, migration, and invasion. The loss of SOCS3 function may be associated with reduced expression due to epigenetic silencing but also with abnormal subcellular localization impairing its function [50]. For these reasons, combined inhibition of the EGFR pathway and important intermediates in redundant crosstalk pathways might be necessary before an improvement of chemotherapeutic effect can be realized [21].
An excessive activation of proliferative pathways should hypothetically result in increased proliferation of cancer cells; however, there appears to be a compromise between maximal mitogenic stimulation and avoidance of antiproliferative defenses such as senescence [1, 51, 52]. In accordance, there are several morphological and metabolic compartments in tumor tissue. The first is the tumor stroma with well-differentiated cancer cells that are nonproliferative and display lower numbers of mitochondria. Secondly, there are less differentiated cancer cells that are highly proliferative (higher Ki-67 expression) and mitochondria-rich [53]. The nonproliferative subgroup of tumor cells is probably positively selected by radiotherapy. In accordance, patients with HNSCC with low proliferation rate (Ki-67 ≤20 %) have a worse response to radiotherapy than patients with high proliferative tumors [54]. High expression of Ki-67 and EGFR suggests that early lymph node metastasis and primary HNSCC tumors have rather high proliferative indexes and therefore, they should be sensitive to radiotherapy [55].
7.2.2 Disruption of Growth-Suppressing Mechanisms
Many tumor suppressors, operating through a variety of mechanisms, have been discovered due to their inactivation in various cancers. Important HNSCC tumor suppressors have also been identified through the effect of human papillomavirus (HPV) infection. Oncogenic HPV subtypes are able to transform epithelial tissue because of the action of the viral oncoproteins E6 and E7 [56]. In particular, the expression of the E6 and E7 leads to the inactivation of two important tumor suppressors, p53 and retinoblastoma protein (Rb). Viral E6 protein stimulates p53 degradation [56] and can also activate telomerase [57]. The E7 protein is able to bind pRb with a preference for the underphosphorylated “active” form of pRb. The Rb proteins play a key role in controlling the checkpoint between the G1 and the S phase of the cell cycle. E7 oncoprotein is also able to inactivate the cyclin-dependent kinase (CDK) inhibitor p21 [15]. HPV status also affects miRNA expression patterns in HNSCC. Certain miRNAs (miR-15a, miR-16, miR-195, miR-497 family, miR-143, mir-145, and miR-106–363 cluster) appear to be crucial for the HPV carcinogenesis [58].
Despite of reduced levels of p53 in HPV-associated tumors (E6-mediated degradation), p53 protein is not mutated and is functional. Radiation-induced p53 production may be sufficient to provoke apoptosis. In contrast, a high percentage of HPV-negative tumors have mutated p53 and create resistance to chemotherapy- and/or radiation-induced apoptosis [59]. Furthermore, promoters of tumor suppressors 14-3-3σ and RASSF1A are hypermethylated, and there are cyclin D gene amplifications in HPV-negative HNSCC more frequently than in HPV-positive tumors [60–62]. Inactivation of 14-3-3σ by promoter hypermethylation promotes metastasis in nasopharyngeal carcinoma [63]; based on the p16 and cyclin D1 expression, patients with downregulated p16 and overexpressed cyclin D1 had worse disease-free and overall survival [64].
Progression through the cell cycle is regulated by the coordinated activities of cyclins and cyclin-dependent kinases (CDK), which are regulated through the binding of CDK inhibitors. The CDK2AP1 (DOC-1) protein is a unique inhibitor of CDK2 and has an important growth-suppressing effect through association with a DNA polymerase α/primase complex [65, 66]. Additionally, CDK2AP1 can induce apoptosis [67, 68]; however, its expression is often reduced in HNSCC [69, 70] and intratumor reduction of CDK2AP1 expression is a negative prognostic marker in patients with surgically resected HNSCC [70]. On the other hand, induction of CDK2AP1 expression in the tumor stroma significantly reduced tumor cell growth and invasiveness [65].
7.2.2.1 TGF-β Signaling Pathway
Comprehensive evidence exists for the deregulation of the TGF-β signaling pathway in tumor formation and advanced disease progression [10, 71]. In normal epithelium, TGF-β suppresses tumor growth through antiproliferative and apoptosis-triggering effects. Nevertheless, as the tumor progresses, autocrine loops of TGF-β are established and tumor cells become resistant to antiproliferative stimuli [72]. Increased TGF-β1 expression is seen in approximately 80 % of HNSCCs and is associated with disease progression and worse survival [8, 73]. TGF-β signaling can activate different proliferative non-Smad pathways including Jnk/p38/MAPK, Ras, Par6, and PI3K/Akt [72, 74], which is in accordance with the fact that increased TGF-β1 expression and activated phosphatidylinositol 3-kinase/Akt signaling are frequently observed as a consequence of disrupted canonical TGF-β signals [75]. Loss of chromosome 18q, a region encoding both Smad2 and Smad4, is common in HNSCC and loss of heterozygosity (LOH) at the Smad4 locus occurs in ~50 % of HNSCC [76, 77]. LOH affecting 18q has been linked to poor survival [78]. Smad4 was downregulated not only in HNSCC malignant lesions but also in normal adjacent mucosa. Furthermore, Smad4 (−/−)-deficient mice developed spontaneous HNSCC [79]. TGF-β promotes the assembly of a cell-surface receptor complex composed of type I (TGFβRI) and type II (TGFβRII) receptor serine/threonine kinases. Low expression of TGFβRII has been detected in a variety of tumors, including HNSCC, and rises as a consequence of genetic alterations or epigenetic silencing [80–82]. In sum, TGFβRII expression may be reduced in >70 % of HNSCCs [10] and correlates with diminished tumor differentiation and more aggressive behavior [83].
miR-211 promotes the progression of head and neck carcinomas directly by targeting TGFβRII [84]. Although genetic alteration of TGFβRI by mutation or deletion is not common in HNSCC, repression of TGFβRI expression via hypermethylation of its promoter has been reported [8, 85], and reduced TGFβRI expression was associated with increased invasion and metastasis in esophageal carcinoma [83]. TGFβRI mutations have been detected in 19 % of HNSCC metastases [86]; however, TGFβRI deletion does not initiate tumor formation in the oral epithelium, but instead allows progression to HNSCC after excessive Ras activation [10, 75]. Taken together, resistance to TGF-β–mediated growth inhibition may arise by different ways [87] inter alia: (1) due to activation of non-Smad pathways triggered by TGF-β, (2) due to mutations in the Smad4 gene [88], (3) due to Smad4 decreased expression, (4) due to increased expression of inhibitory Smad7 [89], (5) due to decreased expression of TGFβRII or TGFβRI, or (6) due to mutations in the TGFβRII or TGFβRI genes.
7.2.2.2 Circadian Clock
Proliferation and the cell cycle regulation are also controlled by the circadian clock [90]. To date, at least nine mammalian core circadian clock genes (CCGs) have been identified: Period1 (PER1), Period2 (PER2), Period3 (PER3), Clock, Cryptochrome1 (CRY1), Cryptochrome2 (CRY2), BMAL1, Caseinkinase1E (CK1E), and Timeless (TIM). CCGs, and the proteins they encode, constitute the circadian oscillator and are responsible for circadian rhythms. Deregulation of the circadian clock may lead to excessive cell proliferation [91]. c-myc activation might be a possible mechanism, as its transcription is directly controlled by circadian regulators [92]. Many studies have reported altered expression of clock genes in tumors [93–96]. Mice deficient in circadian clock genes showed many phenotypic abnormalities, including increased cancer incidence following genotoxic stress [90, 92]. In PER2 mutant mice, expression of proteins involved in tumor suppression and cell cycle regulation (Mdm-2, cyclin D1, cyclin A, Gadd45α, and c-myc) is altered. All nine mentioned CCGs were markedly downregulated in the peripheral blood leukocytes of preoperative HNSCC patients. Restoration of CLOCK and PER1 gene and protein expression were found in postsurgical patients with good prognosis unlike in patients that died within one year after surgery [97]. The expression of CRY1, CRY2, BMAL1, PER1, PER2, PER3, and CK1E genes exhibited significant downregulation in the tumor tissues. Downregulated expression of CRY2, PER3, and BMAL1 was correlated with more advanced cancer stages. Downregulated PER3 and upregulated TIM expression positively correlated with larger tumor size and lower expression of PER3 correlated with deeper tumor invasion. Poor survival was related to lower expression of PER1 and PER3. These results indicate a possible association of circadian clock genes with the pathogenesis and prognosis of HNSCC [98].
7.2.3 Avoiding of Contact Inhibition
In healthy tissues, cells grow inside defined areas and quit growing when they come into contact with the dense extracellular matrix or other cells, a concept referred to as contact inhibition. Neurofibromatosis-2 protein (NF2 or merlin) plays a crucial role in the upkeep of contact inhibition. The growth inhibitory function of NF2 is dependent upon interaction with the cytoplasmic domain of CD44 [99], and numerous studies have indicated that NF2 suppresses tumor growth and metastasis [100, 101]. 22q12.2 is the known locus for the NF2 gene and 22q is a frequent site of loss of heterozygosity (LOH) in HNSCC. Notably, allelic losses on 22q tended to occur especially in patients with stage 3 and 4 disease. Of particular interest is that laryngeal tumors had higher rates of 22q LOH than oral cavity or pharyngeal tumors [102]. The phosphorylated form of NF2 is growth promoting and predominantly appeared in the presence of hyaluronic acid–degrading enzymes (HAases); cells cultured without HAases contained mostly the growth inhibitory form of NF2 [103]. HAases have been found to transform the expression of CD44 isoforms, which could also play a significant role in tumor progression [104]. On the other hand, activity of the HAase Hyal-2 was dependent on the expression of CD44 in both living cells and enzymatic assays [105]. In accordance, salivary soluble CD44 concentration was markedly higher in HNSCC patients compared with healthy controls (1.09 ng/mL for controls and 7.85 ng/mL for HNSCC patients) [106, 107]. Similarly, HAase levels were elevated in HNSCC patients when compared to normal controls [108]. The presence of extremely high molecular weight hyaluronic acid (HMW-HA) and its binding to the CD44 receptor (together with poor HAase) activity played a significant role in the naked mole rat cancer resistance. Furthermore, enzymatic digestion of HMW-HA abrogated contact inhibition of cells [103] and cellular reactions turned on by HA are dependent on the HA polymer length. HMW-HA inhibits proliferation and has anti-inflammatory effects [109] while low molecular weight HAs induce proliferation and inflammation [110] (see Fig. 7.2 and Table 7.1).
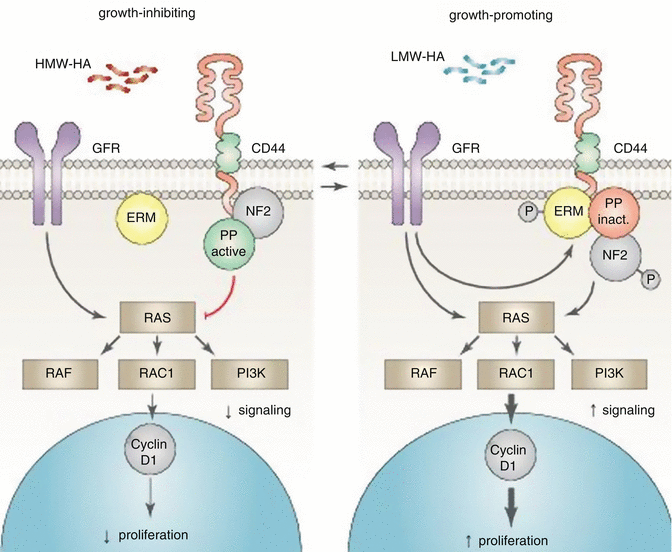
Fig. 7.2
Role of neurofibromatosis-2 protein and hyaluronic acid in contact inhibition. Growth factor receptor (GFR); protein phosphatase (PP) and ezrin-radixin-moesin (ERM) family of band 4.1 proteins. Additional components are likely associated with the CD44-bound complexes. Neurofibromatosis-2 protein (NF2) plays a crucial role in the upkeep of contact inhibition. The growth inhibitory function of NF2 is dependent upon interaction with the cytoplasmic domain of CD44. Furthermore, the phosphorylated (growth-promoting) form of NF2 predominantly appears in the presence of hyaluronic acid–degrading enzymes. The presence of extremely high molecular weight hyaluronic acid (HMW-HA) and its binding to the CD44 receptor plays a significant role in the growth inhibition, while low molecular weight HAs induce proliferation and inflammation (Adapted from Pure et al. [110])
7.3 Markers of Tumor Promotion Inflammation and Oxidative Stress
Inflammatory reactions accompanying early phases of neoplastic transformation create a favorable environment for cancer expansion. The accumulation of growth factors, cytokines, prostaglandins, free radicals, and matrix proteinases triggers pro-tumorigenic mechanisms and influences protein modification, DNA damage, and expression profile changes in genes and miRNAs. Together with the immunosuppressive impact of the adaptive immune response, and interactions between stroma and epithelium, these changes dramatically support tumor expansion [132]. Many tumors, including HNSCC, have been associated with constitutive inflammation and oxidative stress [132–134]. Several studies have reported that the synthesis of metallothionein (MT) was induced during oxidative stress [135–137], and two additional studies discovered increased MT protein levels in HNSCC tumors [138, 139]. The highest MT level was found in the tissues of oral tumors (170 ± 70 μg/g), whereas a relatively modest MT level was found in tumors of oropharynx (130 ± 50 μg/g) [139]. The blood level of MT in healthy controls was much lower than in HNSCC patients [140]. Furthermore, increased immunostaining of MT was also observed in the tissue samples from tonsillar squamous cell carcinoma in comparison with a chronic tonsillitis control group [141]. A positive association between MT protein staining and tumors (vs. healthy tissues) was demonstrated in HNSCC patients (odds ratio, OR, 9.95; 95 % CI 5.82–17.03) [142].
The transcription factor NRF2 manages a cellular response to electrophiles and oxidants by triggering expression of genes important for antioxidant defense. NRF2 is inhibited by Kelch-like ECH-associated protein 1 (KEAP1). Dysregulation of NRF2/KEAP1 pathway seems to be important prior to the formation of HNSCC [143, 144]. NRF2 expression was increased in 91.5 % of HNSCC tumors, and the expression of the NRF2-reguated gene thioredoxin was increased in 75 % of tumors [145].
Nuclear factor-kappa B (NF-ĸB) is a crucial factor in inflammatory and immune pathways that also acts as a tumor promoter [146]. In tumor cells, NF-ĸB can be activated as a result of genetic alterations and can induce the expression of angiogenic factors, inflammatory cytokines, adhesion receptors, or inducible nitric oxide synthase (iNOS) and enzymes involved in the arachidonic acid metabolism such as cyclooxygenase-2 (COX-2). NF-ĸB is also a major survival factor due to its ability to induce expression of Bcl-2 [134]. Long-standing inflammatory signals have been shown to induce an epigenetic switch from healthy cells to cancer through a positive feedback loop containing NF-ĸB, Lin28, let-7, and IL-6 [147, 148]. STAT3, a transcription factor induced by IL-6 (discussed above, section 2.1), is able to activate miR-181b-1 and miR-21 that can trigger the epigenetic switch. MiR-181b-1and miR-21 deactivate the tumor suppressors PTEN and CYLD, respectively, leading to elevated NF-ĸB activity [147]. In HNSCC, a key role of NF-κB in promoting alterations of the proteome and transcriptome has been recently established [149]; and studies have demonstrated NF-κB activation in squamous dysplasia and in tobacco- and viral-associated HNSCC [150]. Increased nuclear levels of NF-κB immunostaining highly correlated with progress of dysplasia and reduced survival in HNSCC patients [151].
Aberrant arachidonic acid (AA) metabolism, especially altered 5-lipoxygenase (5-LOX) and cyclooxygenase (COX), is often involved in HNSCC development [152]. COX-2 and 5-LOX enhance tumor cell proliferation and are proangiogenic due to the induction of FGF, VEGF, and MMPs [153–156]. Cyclooxygenases catalyze the production of prostaglandins (PGs). Elevation of COX-2 has been described at both the mRNA and protein level in HNSCC [157] and was associated with an adverse outcome [158]. Prostaglandin E2 (PgE2) is able to promote proliferation and may also inhibit apoptosis by increasing Bcl-2 expression [159]. Furthermore, PgE2 was shown to elevate the expression of angiogenic factors prior to tumor cell invasion and metastasis [154]. Overexpression of inducible microsomal prostaglandin E synthase (mPGES) elevated the amount of PgE2 in HNSCC cells [160]. Patients with increased COX-2 and PgE2 tumor expression had significantly inferior overall survival [161]. Immunohistochemical experiments showed that an elevation in COX-2 expression was positively correlated with the amount of Foxp3+ regulatory T cells in the microenvironment of HNSCC, indicating that COX-2 enables the expansion of the regulatory T cells [162]. Increased expression of COX-2 resulted in elevated levels of PgE2 and VEGF and enhanced angiogenesis and tumor growth [153]. COX-2 inhibitors reduced invasion and viability of HNSCC cells by downregulating VEGF, MMP-2, and MMP-9 secretion [159, 163, 164]. Taken together, inflammatory mediators such as ROS, metabolites of arachidonic acid, and cytokines contribute to tumor progression by inducing oncogenic mutations, resistance to apoptosis, adaptive responses, and changes in the microenvironment, including induction of angiogenesis.
7.4 Avoiding Immune Destruction
The immune system acts as an important barrier against tumorigenesis and tumor progression.
The unaffected immune system can eliminate cancer cells, but the effectiveness of immune antitumor defense is often suppressed by the tumor microenvironment. There is significant evidence that reactivation of an antitumor immune response can cause tumor regression and have a favorable clinical impact [165]. Tumor cells generate high levels of proinflammatory agents (such as ROS); nevertheless, the tumor microenvironment sides with immune suppressors rather than with immune effectors [165]. For example, soluble phosphatidylserine (sPS) released from tumor cells is able to interact with the PS receptor on macrophages to trigger an anti-inflammatory response and immune escape [166]. The tumor immune infiltrate is also shifted toward an anti-inflammatory status and mode of immunosuppression, due to the expression of surface molecules such as PD-L1 and reduced expression of molecules that present antigens [167].
7.4.1 Immune Cells
HNSCC patients typically display significant immunosuppression. They have elevated Treg (CD4+ CD25+ FoxP3+ regulatory T cells) and increased progenitor CD34+ cells that are able to suppress CD4+ helper T cells and CD8+ cytotoxic T cells at the primary tumor area. Treg presence was associated with a poor prognosis [168–170]. Treg can inhibit T-cell reactivity and help to expand the level of inhibitory cells by converting CD4+/CD25- cells into inhibitory CD4+/CD25+/FoxP3+ cells [171]. It also appears that an enrichment in circulating Treg is a long-lasting immune modification that is not overturned by cancer therapy [172]. Cancer patients often display an increased neutrophil/lymphocyte ratio, a low neutrophil and CD14 (high) CD16+ monocyte activation state, and an elevated CD4/CD8 ratio, which has been related to poor survival. In contrast, a high percentage of CD98+ Th cells appeared to be associated with a better outcome [173].
Natural killer (NK) cells are crucial for the management of immune response and removal of faulty cells with downregulation of MHC I molecules. In HNSCC, NK cells appear to be strongly defective. The habitual downregulation of MHC I molecules provides the tumor cells invisibility to T-cell–mediated immunity; NK cells seemingly evolved as an evolutionary response to the downregulation of MHC I molecules [174]. Nevertheless, recent studies have demonstrated that tumor cells are able to internalize NK cells. Survey from long-term real-time imaging experiments indicates that almost all of the NK cells (>95 %) die within 24 h after internalization [175]. Furthermore, internalization of T cells by melanoma cells provides a survival advantage to the tumor cells under conditions of starvation [176]. Internalization of NK cells into tumor cells requires the actin cytoskeletal regulator (ezrin) and leads to programmed cell-in-cell death of NK cells [175]. Patients with tumors expressing high ezrin levels had shorter disease-free survival than those with low expression [177]. NK cells can be stimulated by double-stranded RNA through Toll-like receptor 3 (TLR3). TLR3 was expressed on the cell surface of naive NK cells but was quickly internalized due to the action of the HNSCC microenvironment. This TLR3 internalization presents other possible immune evasion mechanism in HNSCC [178].
7.4.2 Surface Molecules
Host immune recognition is avoided in HNSCC by several mechanisms, including reduced expression of MHC classes I and II, and upregulation of programmed cell death ligand-1 (PD-L1) and FasL [179–182]. The expression of MHC class I molecules on the cell surface is necessary for the presentation of peptide antigens to cytotoxic CD8+ T lymphocytes. Approximately 50 % of HNSCC cases demonstrated a loss of class I HLA molecules that could be correlated with regional lymph node metastasis [181]. Downregulation of class I major histocompatibility complex (MHC) molecules shields tumor cells from immune recognition. In addition, upregulation of FasL leads to apoptosis of T cells. Recent studies have shown that plasma of patients with oral cancer contains FasL+ membranous vesicles having the capacity to induce T-cell death [183, 184]. The transfer of activated T cells after blocking PD-L1 with a neutralizing antibody (10B5) delayed the development of HNSCC and resulted in enhanced survival, compared with the transfer of T cells alone [167].
7.4.3 Immune Regulators
One of the mechanisms by which HNSCC tumors can weaken host immune reactions is through the alteration of the cytokine environment at the tumor area. Recent studies have shown that alteration of cytokine immune responses can promote growth and metastasis [185]. The production of cytokines such as IL-6 and IL-10 by tumor cells supports a Th2 response that is accompanied by a substantially weakened antitumor defense [184, 186]. As a consequence of the shift from the Th1 to the Th2 type of T-cell cytokine response, HNSCC tumors produce elevated levels of immunosuppressive agents such as TGF-β that can directly inhibit cytotoxic T cells and NK cell–mediated immunity [187] and contribute to the recruitment of immunosuppressive M2 macrophages and myeloid-derived suppressor cells (MDSCs) [185, 188, 189]. In summary, TGF-β mutes antitumor immunity while promoting tumor-supporting inflammation.
Granulocyte-macrophage colony-stimulating factor (GM-CSF) and PgE2 usually have proinflammatory effects that promote the maturation of neutrophils and macrophages in the initial phases of inflammation. They are also secreted by the HNSCC tumor cells to favor angiogenesis, growth, and immune response avoidance [39]. Tumor-produced GM-CSF has been found to stimulate MDSC maturation and recruitment, and increased levels of GM-CSF are related to an adverse prognosis in HNSCC patients [169, 190]. Elevated levels of PgE2 are associated with angiogenesis and invasiveness in aggressive primary tumors [191] and PgE2 expression is associated with decreased CD8+ T-cell numbers and increased immune suppressor cells at the tumor area. PgE2 can induce the secretion of IL-10 and repress the secretion of cytokines by CD4+ T cells directly, and PgE2 seems to be crucial for tumor-associated immunosuppression [184–186]. Monocyte chemotactic protein 1 (MCP-1), produced by tumor cells, attracts IL-10– and TGF-β–secreting M2 macrophages leading to increased immunosuppression [192]. The maturation of dendritic cells in patients with HNSCC seems to be impaired by tumor-produced VEGF [181, 193, 194]. Furthermore, within HNSCC tissue, plasmacytoid dendritic cells have been shown to be defective in their capacity to produce interferon alpha (IFN-α), a cytokine that is important for antitumor reactivity [195]. A significant decrease in serum IL-13, IFN-γ, and MIP-1β levels, and a significant increase of serum inducible protein-10 (IP-10), in HNSCC patients was demonstrated, regardless of primary tumor site [196]. Neutrophil/lymphocyte ratio and serum concentrations of IL-8, CCL4 (MIP-1β), and CCL5 (RANTES) were significantly higher in the peripheral blood of HNSCC patients than controls [197].
Main HNSCC protective mechanisms against immune reactivity are shown in Table 7.2.
Table 7.1
Markers of chronic proliferation in HNSCC
|
Biomarker
|
Mechanism
|
Detected in
|
References
|
|---|---|---|---|
|
c-Met
|
↑ protein expression
|
Tumor tissue
|
[111]
|
|
CEP55
|
↑ mRNA and protein expression
|
Tumor tissue
|
[112]
|
|
CCGs
|
↓ mRNA and protein expression
|
Peripheral blood leukocytes, tumor tissue
|
|
|
CDK2AP1
|
↓ mRNA and protein expression
|
Tumor tissue
|
|
|
CD44
|
↑ protein expression
|
Saliva
|
|
|
Cyclin D1
|
↑ protein expression, gene amplification
|
Tumor tissue, HNSCC cell lines
|
|
|
DST
|
↓ mRNA expression
|
Tumor tissue
|
[118]
|
|
MAGE
|
↑ mRNA expression
|
Saliva
|
[119]
|
|
MYCN
|
↑ mRNA expression
|
Tumor tissue
|
[118]
|
|
TGF-α
|
↑ mRNA and protein expression
|
Tumor tissue, HNSCC cell lines
|
|
|
TGF-βI
|
↑ protein expression
|
Plasma
|
[83]
|
|
RASSF1A
|
↓ mRNA and protein expression, LOH
|
Tumor tissue, HNSCC cell lines
|
|
|
14-3-3σ
|
↓ mRNA and protein expression
|
Tumor tissue, HNSCC cell lines
|
[63]
|
|
EGFR
|
↑ mRNA and protein expression
|
Tumor tissue, HNSCC cell lines
|
|
|
HAase
|
↑ mRNA and protein expression
|
Saliva, HNSCC cell lines
|
[108]
|
|
Ras
|
↑ mRNA and protein expression, gain-of-function mutations
|
Tumor tissue
|
|
|
PI3KCA
|
↑ protein expression, gene amplification
|
Tumor tissue
|
|
|
IL-8
|
↑ protein expression
|
Saliva, serum
|
|
|
p53
|
Inactivation, mutation, LOH
|
Tumor tissue, surgical margins
|
|
|
NF2
|
↓ mRNA and protein expression, 22qLOH
|
Nf2-deficient tumor-bearing mice, blood
|
|
|
IL-6
|
↑ mRNA and protein expression
|
Saliva, serum, HNSCC cell lines, tumor tissue
|
|
|
TNF-α
|
↑ protein expression
|
Saliva
|
[43]
|
|
PTEN
|
↓ protein expression, LOH
|
Tumor tissue
|
|
|
CDK4
|
↑ protein expression
|
Tumor tissue
|
[115]
|
|
pRb
|
↓ protein expression, inactivation, LOH
|
Tumor tissue
|
|
|
SET
|
↑ protein expression
|
Tumor tissue, HNSCC cell lines
|
|
|
SMAD4
|
↓ protein expression, LOH, inactivation
|
HNSCC cell lines, tumor tissue, SMAD4-deficient tumor-bearing mice
|
|
|
STAT3
|
↑ protein expression, activation
|
Tumor tissue
|
[35]
|
|
SOCS1 and SOCS3
|
↓ protein expression, mRNA expression, epigenetic silencing, abnormal subcellular localization
|
Tumor tissue, HNSCC cell lines
|
|
|
TGFβRI
|
↓ protein expression, epigenetic silencing
|
Tumor tissue
|
|
|
TGFβRII
|
↓ protein expression, epigenetic silencing, mutation
|
Tumor tissue, HNSCC cell lines
|
|
|
VEGF
|
↑ protein expression
|
Tumor tissue, HNSCC cell lines, serum
|
[39]
|
|
AKT2
|
↑ protein expression, gene amplification
|
Tumor tissue
|
[5]
|
|
EBV
|
A causative agent for most nasopharyngeal carcinomas; plasma EBV DNA load is an independent prognostic factor
|
Tumor tissue, blood
|
|
|
HPV
|
A causative agent for most oropharyngeal cancers
|
Tumor tissue, blood
|
Table 7.2
HNSCC protective mechanisms against immune reactivity
|
Mechanism
|
Biomarker
|
Consequence
|
References
|
|---|---|---|---|
|
Escape from immune recognition
|
Loss of MHC
|
↓ presentation of tumor antigens
|
|
|
↑ PD-L1 expression
|
↑ T-cell apoptosis
|
||
|
↑ FasL expression
|
↑ T-cell apoptosis via Fas-FasL
|
||
|
↑ ezrin expression
|
↑ internalization of NK cells, NK cell apoptosis
|
[175]
|
|
|
Internalized TLR3
|
NK cell impairment
|
[178]
|
|
|
Modulation of the cytokine response
|
↑ PgE2, COX-2
|
Decreased levels of CD8+ T cells; expansion of the regulatory T cells (Treg)
|
|
|
↑ TGF-β
|
Inhibition of cytotoxic T cells and NKs; MDSCs and M2-skewed macrophages recruitment
|
||
|
↑ IL-10
|
M2-skewed macrophage activation and promotion of a Th2 response
|
||
|
↑ VEGF
|
Impaired maturation of dendritic cells
|
||
|
↑ GM-CSF
|
MDSCs recruitment
|
[39]
|
|
|
↓ IFN-α and IFN-γ
|
↓ antitumor reactivity
|
||
|
Induction of immune suppressor cells
|
MDSCs recruitment
|
Suppression of T-cell proliferation and activity
|
|
|
M2-skewed macrophages recruitment
|
Downregulation of T-cell activity and secretion of high levels of growth factors, angiogenic factors, and matrix remodeling enzymes
|
[192]
|
|
|
Treg (CD4+ CD25+ FoxP3+ regulatory T cells) expansion
|
Suppression of CD8+ T cell– and CD4+ helper T cell–mediated immunity
|
||
|
CD34+ progenitor cell expansion
|
Suppression of CD8+ T-cell– and CD4+ helper T-cell–mediated immunity
|
7.5 Markers of Tumorous Angiogenesis
Angiogenesis is an essential process for tumor progression, outgrowth, and dispersion. Vascular endothelial growth factor A (VEGF-A) is a well-characterized agent that can induce angiogenesis. VEGF-A expression can be upregulated by hypoxia, through the action of hypoxia-inducible factor 1 alpha (HIF-1α), and by oncogene signaling through EGFR, Raf, MEK, and PI3K signals [198, 199]. von Hippel-Lindau (VHL) is a tumor suppressor gene located on chromosome 3p that is involved in a multiprotein complex that regulates oxygen-dependent degradation of HIF-1α [199]. Loss of heterozygosity (LOH) within VHL was found at a high frequency (45.5 %) in tongue squamous cell carcinoma patients [200]. Loss of VHL function decreases HIF-1α proteolysis under aerobic conditions and thereby elevates expression of many HIF-1α downstream genes [201]. Furthermore, osteopontin (OPN) expression correlates negatively with VHL expression, suggesting OPN connection with tumor hypoxia. Accordingly, OPN expression was associated with HNSCC tumor progression [202, 203]. Overexpression of VEGF (in tumor tissue or in serum of HNSCC patients) as a result of hypoxia or oncogene signaling was correlated with resistance to cytotoxic treatment, worse prognosis, and advanced disease [204–208]. In a meta-analysis of 1002 HNSCC patients, VEGF was significantly associated with decreased survival [209].
There are many other factors playing important role in angiogenesis, including platelet-derived growth factor (PDGF), epidermal growth factor (EGF), hepatocyte growth factor (HGF), prostaglandins, TGF-β, COX-2, and IL-6 [10, 36, 73, 153, 154, 210]. Oral cancer overexpressed 1 and 2 (ORAOV1 and 2) are transmembrane proteins involved in tumor angiogenesis and regulation of cell growth via VEGF induction [211, 212]. Overexpression of ORAOV1 as well as ORAOV2 was reported in HNSCC [212, 213]. Loss of transforming growth factor-beta type II receptor (TGFβRII) is commonly observed in human HNSCC and leads to increased inflammation and angiogenesis [10].
Fibroblast growth factors (FGF) provide another angiogenic signaling mechanism in the tumor microenvironment. Basic fibroblast growth factor (bFGF) is a potent inducer of angiogenesis and is usually found immobilized on the extracellular matrix within the tumor milieu. Expression of the secreted binding protein for fibroblast growth factors (FGF-BP) appears to be a mechanism by which immobilized FGF can be activated [214, 215]. FGF-BP is produced extensively by primary HNSCC tumors and some metastatic cells but not in normal adult mucosa [216].
Hasina et al. studied the expression of cytokines FGF-2, VEGF, IL-8, and HGF in HNSCC, dysplastic, and healthy samples and demonstrated different ways by which particular tumors can provoke angiogenesis [217]. The highest expression of all studied cytokines was in HNSCC tissues, and two distinct tumor clusters were identified with regard to angiogenesis. One group of tumors had significantly increased microvessel density with high levels of FGF-2 and VEGF and relatively low levels of IL-8 and HGF. The second group of tumors displayed lower microvessel density and expressed low levels of FGF-2 and VEGF and higher levels of IL-8 and HGF. This might have some relevant therapeutic consequences, because it is not rational to inhibit angiogenesis by targeting molecules that are poorly expressed in treated type of tumor.
NF-κB is also vastly important for angiogenesis in tumors. Some NF-κB downstream genes are powerful angiogenic agents, such as VEGF, IL-8, and COX-2 [134, 218]. Thrombospondin-1 (TSP-1) was recently identified as a target gene of Ras-modulated Myc activity, which represses TSP-1 expression and increases tumor angiogenesis. In HNSCC patients, TSP-1 was found to be downregulated [219, 220].
Thus far, clinically tested antiangiogenic therapies have unfortunately demonstrated very disappointing effectiveness, primarily in regard to overall survival. Several studies have reported that drugs targeting VEGF can suppress the growth of primary tumors but may also promote tumor metastasis [221, 222]. Furthermore, anti-VEGF agents induced the production of several cytokines (granulocyte colony–stimulating factor, placental growth factor, osteopontin, IL-6, erythropoietin) that may promote VEGF-independent angiogenesis and metastasis [223]. Another possible unwanted side effect of VEGF inhibitors is suppression of pericytes on tumor vessels. As a result, leaky and immature vessels facilitate the entry of tumor cells and consequent metastatic spread [224].
7.6 Genomic Instability and HNSCC Progression
Progression of HNSCC is usually associated with vast chromosomal abnormalities and subsequent alterations in gene expression. Recently, the presence of more subtle expression regulators such as microRNAs was discovered.
7.6.1 MicroRNA (miRNA)
Many dysregulated miRNAs have been identified that contribute to HNSCC progression. Microarray analysis disclosed ten miRNAs that could differentiate malignant head and neck cancer samples from paired normal tissues; seven miRNAs (miR-29b-1*, miR-181a, miR-181a-2*, miR-181b, miR-221*, miR-744, and miR-1271) were upregulated in cancer samples while three miRNAs (miR-95, miR-101, and miR-141) were downregulated [225]. Some miRNAs seem to be related to the progression of carcinoma or premalignant lesion in head and neck malignancies. miR-21, miR-181b, and miR-345 expression were positively correlated with increased lesion severity [226] and miR-137 and miR-193a were reported to be tumor suppressors that were often epigenetically silenced during oral carcinogenesis [227]. With the exception of only a few miRNAs, all reported HNSCC miRNA studies to date have shared little in common; thus, the exact importance/contribution of particular miRNA species remains to be elucidated. Upregulation of miR-21, miR-31, miR-10b, miR-181, and miR-221 and downregulation of miR-133a/b, miR-138, miR-139, miR-375, and miR-200c are, however, trends that have been consistently identified in HNSCC [228–232]. Increased expression of miR-21, miR-18a, and miR-221 and decreased miR-375 and the ratio of miR-221/miR-375 expression were found to have a high specificity and sensitivity for diagnosis of HNSCC [233].
7.6.2 Chromosomal Aberrations
Chromosomal rearrangements resulting in very complex karyotypes are often found in HNSCC; in fact, more than 70 % of tumors are aneuploid [234]. Several genes associated with the DNA damage response pathway are frequently altered in HNSCC, including BRCA1, BRCA2, FANCD2, and FANCG. Other cancer-related genes linked to hereditary cancer syndromes such as VHL, MLH1, XPC, RB1, [235], and recently discovered germline alterations in RAD51C, are also known to be mutated in HNSCC [236]. SMAD 4 deficiency (loss of chromosome 18q) is a common mutation that suppresses Fanc-/Brca-mediated DNA repair and increases genomic instability [8]; and recent findings suggest that interactions with tobacco and polymorphisms in XRCC1 and XRCC2 could increase the risk of HNSCC [237–239]. Furthermore, viral E7 protein is able to disrupt centrosome duplication causing abnormal mitosis, aneuploidy, and increased genomic instability [240]. NK cell internalization is another mechanism that results in aneuploidy. It is hypothesized that NK cell internalization might facilitate the development of aneuploidy in tumor cells by interfering with cell division resulting in disturbed chromosome segregation and/or cytokinesis [175]. To date, more than 30 sites of significant chromosomal aberrations and 15 frequently mutated genes including TP53, CDKN2A, HRAS, PIK3CA, NFE2L, and NOTCH1 have been discovered in HNSCC patients. Most of these genetic alterations occur also in lung squamous cell carcinoma [144, 241]. Genomic regions most frequently amplified (>35 %) were located on 3q (candidate genes AIS (p40/p73L), SSR3, CCLN1, hTERC, SKIL, ECT2, PIK3CA, SST, TP63, and TFRC) [242, 243]), 5p, 8q (candidate gene Myc), 9q, 11q (candidate gene CCND1), 20q, and 7p (candidate gene EGFR on 7p12); 7p and 3q amplification occurs in early tumor development [234, 235, 244]. Regions most frequently deleted (>40 %) involved chromosomes 3p (FHIT gene maps to 3p14.2, VHL to 3p25, RASSF1A to 3p21), 8p, 9p (candidate genes p16/CDKN2A), 13q, and 18q [235, 244]. The loss of 18q is associated with a worse clinical outcome [245]. Rearrangements that often occur in higher grades of dysplasia and SCC include PTEN (10q23.3) inactivation, 11q13 amplification, and LOH at 6p, 8p, 13q21, 14q32, and 4q27 [246–249]. Amplification of 11q13 was found in about one-third of HNSCC, with amplified cyclin D1 playing crucial role in promoting the cancer phenotype [250, 251]. Gain on chromosome 8q22.3, the location of YWHAZ (14-3-3 zeta), is found in 30–40 % HNSCC cases [252]. LOH analyses suggest that initial alterations target crucial genes located on chromosomes 3p (RASSF1A and VHL), 9p21 (cyclin-dependent kinase inhibitors), and 17p13 (TP53) [200, 253] (Fig. 7.3). LOH of the 9p21 region occurs in 70 % of dysplastic lesions in oral tissues simultaneously with promoter hypermethylation; subsequent inactivations of the remaining alleles of 14 ARF and p16 are crucial events in HNSCC progression [126, 248].
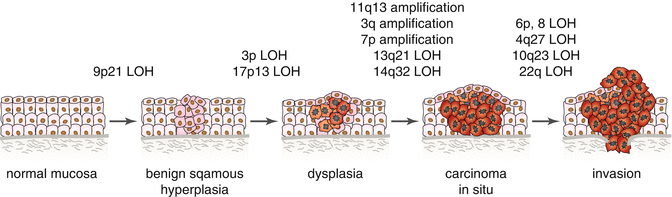
Fig. 7.3
Genetic changes associated with the histologic progression of HNSCC based on changes of chromosomal material. Loss of heterozygosity (LOH); candidate tumor suppressor genes include p16 (9p21), p53 (17p13), RASSF1A (3p21), FHIT (3p14.2), VHL (3p25), PTEN (10q23), NF2 (22q12.2), and Rb (13q21). Candidate proto-oncogenes include CCND1 (11q13), CCLN1 (3q25.31), EGFR (7p12), PIK3CA (3q26.31-26.32), AIS (3q), and ORAOV1 (11q13) (Adapted from Polanska et al. [15])
Some studies revealed that distinct chromosomal aberrations can be connected with clinical parameters such as losses at 11p13-p14, 10q23-q26, 11q14-24, 17q11, 8p, 8q, and 6p and gains at 4q11-22 and Xq12-28 that are significant for metastatic carcinoma [118, 234, 248, 249]. Some genes involved in cell signaling (NGFRAP1L1, GPRASP2, BEX1, BEX2, and ZNF6) reside on chromosome Xq21–22, whose amplification is associated with HNSCC metastasis [118]. This could possibly help explain why the female sex is associated with a bad response to organ-sparing therapy and poor outcome [120]. Furthermore, LOH affecting 18q, 22q, and 3p25 and amplification at 14q23-q24.2 (HIF-1α gene) and chromosomal gains on 1q and 16q have already been linked to poor survival [78, 102, 200, 245, 254, 255].
7.6.3 HNSCC Subtypes
With regard to the presence of known genetic alterations occurring in tumors, HNSCC can be divided into four subtypes, which will be discussed below [144].
7.6.3.1 Classical Subtype (CL)
Comprising ~18 % of HNSCC, CL is also found in lung squamous cell cancer and is associated with alterations of the oxidative stress genes KEAP1, CUL3, and NFE2L2 [143, 144, 256]. This subtype manifests genomic alterations typical for SCC (deletion of 9p and 3p, amplification of 3q, and gene amplification of CCND1 and EGFR). The CL subtype is also associated with heavy smoking and exhibits elevated expression of genes induced by cigarette smoke exposure, including NFE2L2, GPX2, and AKR1C1/3 [144]. In particular, TP53 mutation, CDKN2A loss of function, and larynx subsite co-occurred in most classical subtype tumors [256].
7.6.3.2 Atypical Subtype (AT)
About 24 % of HNSCC is AT. This subtype demonstrates no amplification of EGFR, is HPV-positive, and expresses high levels of PI3 kinase (PIK3CA); the name atypical was used due to the lack of EGFR amplification or 9p deletion. AT tumors show a clear HPV+ signature represented by increased expression of transcription factors such as RPA2, CDKN2A (p16), and LIG1 [144, 257]. Additionally, the atypical subtype is characterized by an occurrence of activating mutations in PIK3CA [256].
7.6.3.3 Mesenchymal Subtype (MS)
MS accounts for ~27 % of HNSCC and mostly contains mutations in FGFR1 and FGFR2. The name mesenchymal was chosen because of the significant expression of epithelial to mesenchymal transition (EMT) markers (VIM, DES, TWIST1, and the growth factor HGF) observed in these tumors [144, 258]. MS tumors typically have alterations in innate immunity genes, inter alia high expression of CD56 (NK cell marker) [256].
7.6.3.4 Basal Subtype (BA)
Thirty-one percent of HNSCC are of the BA subtype, which appear similar to basal epithelial cells in airways for which they were named. BA cells have a characteristic gene expression pattern inclusive of higher expression of EGFR, COL17A1, and the transcription factor TP63. An inactivation of NOTCH1 with intact oxidative stress pathways and rather rare alterations of chromosome 3q are typical for the BA subtype. Moreover, the basal subtype included tumors with co-mutations of the HRAS and CASP8 and with 11q13 and 11q22 co-amplification [256].
The CL and AT subtypes demonstrate higher expression of ALDH1 and SOX2 compared to MS and BA. BA seems to express less SOX2 than healthy tonsil tissue [256]. Both of these genes have been hypothesized to be cancer stem cell markers due to their role in self-renewal and pluripotency [259, 260]. The BA subtype expressed significantly more TP63 than any other subtype. Patients in the HPV-negative AT subgroup generally have the most adverse outcome [144].
7.7 Replicative Immortality and Stemness
An intrinsic cellular mechanism allows normal cells to divide only a finite number of times and blocks cell division beyond a certain limit. Limited telomerase activity usually results in telomere erosion and activation of proliferative barriers (senescence and crisis/apoptosis) [1]. The ability of cancer to escape these barriers is due to several mechanisms; reversible polyploidy [2], CDK1 or survivin overexpression, p53 inactivity, and telomerase activation have been well characterized [1, 261–263]. Telomerase activation can be caused inter alia by the HPV-16 viral E6 protein [57]. It has been hypothesized that telomeres are involved in unrestricted proliferation by preventing erosion of the chromosomes [263]. In accordance, the levels of telomerase activity in HNSCC tissues are significantly higher than those in the normal tissues [264] and expression of telomerase in saliva of the oral cancer patients was also increased [265]. Furthermore, Mao et al. detected telomerase activity in 100 % of examined HNSCC cell lines, in 90 % of invasive tumors, in 100 % of dysplastic lesions, and in 100 % of examined hyperplastic lesions, whereas no normal tissues or hyperkeratotic lesions had detectable telomerase activity [266]. Multivariate analysis revealed that overall stage, tumor depth, and telomerase activity were independent variables associated with poor survival [267]. Telomerase expression in peripheral blood mononuclear cells (PBMCs) was correlated with N stage and the prognosis of HNSCC patients [204]. At the RNA level, miR-21, let-7, miR-107, miR-200c, and miR-138 appear to have complex roles in the modulation of immortality, stemness, and epithelial-mesenchymal transition (EMT) in HNSCC [230]. HNSCC tumor cells are morphologically and functionally heterogeneous which implies they arise from specific progenitor cells and not merely from the clonal expansion of a single mutated cell with telomerase activation [268]. Substantial evidence has shown that a small subpopulation of cancer stem cells (CSC) is responsible for tumor initiation and progression. CSCs are described as a small percentage of cells in a tumor that are able to self-renew and produce daughter cells. CSCs are associated with a specific state of differentiation (e.g., mesenchymal features) [269, 270]. EMT is a process by which a polarized epithelial cell develops a mesenchymal phenotype, and it appears to be involved in the process leading to the acquisition of stemness by epithelial tumor cells [271]. CSCs are highly important for disease prognosis due to their crucial role in recurrence and metastasis. Possible biomarkers associated with CSCs in HNSCC are summarized in Table 7.3. All mentioned stemness markers are usually associated with bad prognosis.
Table 7.3
Biomarkers of stemness
|
Biomarker
|
Mechanism
|
Detected in
|
References
|
|---|---|---|---|
|
ABCG2
|
↑ mRNA and protein expression
|
CSC-like subpopulation of tumor cells
|
[272]
|
|
ALDH1
|
↑ protein expression
|
CSC-like subpopulation of tumor cells
|
|
|
CD44
|
↑ mRNA and protein expression
|
CSC-like subpopulation of tumor cells, peripheral blood
|
|
|
Oct4
|
↑ mRNA and protein expression
|
Tumor tissue, HNSCC cell lines
|
|
|
Sox
|
↑ mRNA expression
|
CSC-like subpopulation of tumor cells
|
[269]
|
|
Nanog
|
↑ mRNA expression
|
CSC-like subpopulation of tumor cells
|
[269]
|
|
CD133
|
↑ protein expression
|
CSC-like subpopulation of tumor cells
|
|
|
Telomerase
|
↑ protein expression, activation
|
CSC-like subpopulation of tumor cells
|
[2]
|
|
HMGA2
|
↑ mRNA and protein expression
|
Tumor tissue, CSC-like subpopulation of tumor cells
|
[268]
|
|
ZEB1/2
|
↑ expression
|
Tumor tissue, CSC-like subpopulation of tumor cells
|
[283]
|
|
ADAM17
|
↑ expression
|
Tumor tissue, HNSCC cell line
|
|
|
BMI-1
|
↑ mRNA and protein expression
|
Tumor tissue, CSC-like subpopulation of tumor cells
|
|
|
c-Met
|
↑ protein expression
|
Tumor tissue, CSC-like subpopulation of tumor cells
|
|
|
miR-200c
|
↓ expression
|
Tumor tissue, CSC-like subpopulation of tumor cells
|
|
|
Beta-catenin
|
↑ protein expression
|
Primary HNSCC cell line, CSC-like subpopulation of tumor cells
|
[280]
|
|
GRP78
|
↑ protein expression
|
Tumor tissue, CSC-like subpopulation of tumor cells
|
[290]
|
|
LGR5
(also known as GPR49)
|
↑ mRNA and protein expression
|
Tumor tissue, HNSCC cell lines
|
[291]
|
|
STAT3
|
↑ mRNA and protein expression
|
Tumor tissue, mouse embryonic stem cells, epiblast stem cells
|
|
|
S100A4
|
↑ mRNA and protein expression
|
Tumor tissue, CSC-like subpopulation of tumor cells, HNSCC cell line
|
[294]
|
7.8 Invasion and Metastasis
Many studies have investigated variances in expression profiles of primary and metastatic HNSCC and discovered that specific gene expression changes are required for metastasis [295]. One of the key processes facilitating metastasis is EMT, the process by which a polarized epithelial cell develops a mesenchymal phenotype. EMT is associated with elevation of invasiveness, recurrence, and a worse prognosis in many cancers, including HNSCC. During EMT, low E-cadherin expression and increased vimentin are common [271, 296]. E-cadherin transcriptional repressors such as Snail (SNAI1), Slug (SNAI2), ZEB-1/2, SIP-1, E12/E47 [297, 298], and Twist [299] have traditionally been implicated in promoting EMT. Slug was extremely elevated in the HNSCC cells in response to hypoxia [300]. Smad2 loss can increase EMT; the absence of Smad2 can increase Smad3/4 binding at the Snail promoter thereby increasing Snail transcription and triggering EMT [8]. Finally, Bmi-1 also regulates Snail expression and promotes metastasis [301].
Dysregulation of miRNAs has also been reported during EMT and metastasis. For example, Twist can activate the expression of miR‐10b [302] promoting migration and invasion [230, 303], an association between cancer metastasis and miR-211 expression in OSCC is known, and miR-211 expression was increased in tumors with vascular invasion and correlated with poor prognosis [84, 304]. Overexpression of miR-181 was associated with vascular invasion, lymph node metastasis, and worsened survival rates. Ectopic expression of miR-181 promoted invasion and cell migration but did not apparently promote nor support anchorage-independent growth of OSCC cells. Increased miR-181 levels were found in both tumor tissues and plasma [305]. Oncogenic miR-31 was found to increase HIF1-α expression and tumorigenicity in HNSCC [306]. Reduced miR-138 expression was found in highly metastatic cells. Renewed expression of miR-138 reduced invasion, caused cell cycle arrest, and promoted apoptosis [307, 308]. MiR-138 has been reported to modulate invasivity and migration through targeting the Ras homolog gene family, particularly RhoC and Rho-associated kinase ROCK2. Restricted miR-138 expression may cause elongated, spindle-shape cell morphology, enhance cell stress fiber production, and increase cell migration and invasion [309]. miR-34c is another suppressor of metastasis that targets c-Met to inhibit cell growth and invasivity [310]. miR-203 inhibits HNSCC lung metastasis by repression of pro-metastatic factors affecting extracellular matrix remodeling (SPARC), cytoskeletal dynamics (LASP1), and cellular metabolism (NUAK1). Expression of miR-203 and its signaling network corresponds with HNSCC overall survival [311]. Results also indicated that stem renewal factor Bmi-1 is a direct target of inhibition for miR-203 [312]. Furthermore, transfected miR-133a, miR-133b, and miR-145 (tumor suppressor miRNAs) inhibited cell proliferation and cell invasion in esophageal squamous cell carcinoma (ESCC) cells. miRNAs with binding sequences in the 3′UTR of fascin homolog 1 (FSCN1, actin-binding protein) decreased oncogenic FSCN1 expression [313]. Renewed expression of miR-141 and miR-200c significantly decreased the migration potential of HNSCC cells [314]. Salivary miR-31 is elevated in OSCC and may serve as a useful predictor for early detection and postoperative follow-up [315], while miR-200a and miR-125a are significantly under-expressed in the saliva of OSCC patients [316]. In plasma, miR-31 [317], miR-10b [318], miR-24 [319], miR-181 [305], and miR-184 [320] are upregulated in OSCC patients.
EMT appears to be involved in the process leading to CSC’s occurrence; hence, the biomarkers associated with stemness (Table 7.3) can also serve as metastasis markers. For example, elevated expression of c-Met supports invasion and metastasis of oral tongue carcinoma [321, 322]. Other crucial metastasis markers are shown in Table 7.4. In high-grade HNSCC, dysregulation of E-cadherin and NF-κB signaling can be found together with loss of tight and adherens junctions. Subsequent transformation of cells to a spindle shape enables movement through the basal membrane and promotes metastasis [323, 324]. Masood et al. showed that ALDH1A3, CAV-1, MMP-7, OCT-4, TRIM-29, and TLR-4 proteins have elevated expression in HNSCC cells with severe metastatic phenotypes suggesting that these proteins could have a crucial importance in the metastatic potential of HNSCC cells [55]. Rickman et al. compiled a list of metastasis predictor genes in a four-gene model (FLOT2, HSD17B12, KRT17, and PSMD10) that excluded HPV-positive samples. The four-gene model was highly predictive for expansion of metastasis (hazard ratio 6.5; 95 % confidence interval 2.4–18.1) and predicted occurrence of metastasis with 74 % sensitivity and 78 % specificity [118]. Furthermore, it was reported that tumors with developed metastases had decreased expression of genes associated with apoptosis (PPP2R1B, CASP1, IL18, DAPK3), negative cell cycle regulation (DST) and cell interactions (COL17A1), and increased expression of signaling genes such as MYCN and LRP6.
Table 7.4
Biomarkers involved in metastasis
|
Biomarker
|
Function
|
Expression
|
Detected in
|
References
|
|---|---|---|---|---|
|
ALDH1A3
|
Aldehyde dehydrogenase
|
↑ mRNA, protein
|
Tumor tissue, HNSCC cell lines
|
[55]
|
|
CASP1
|
Apoptosis
|
↓ mRNA
|
Tumor tissue
|
[118]
|
|
CAV-1
|
Signal transduction, vesicular transport, and cell receptor localization
|
↑ mRNA, protein
|
Tumor tissue, HNSCC cell lines
|
|
|
CCR7
|
Chemokine receptor
|
↑ mRNA, protein
|
Tumor tissue, HNSCC cell lines
|
|
|
c-FLIP
|
Procaspase-8-like regulator of death ligand–induced apoptosis
|
↑ mRNA, protein
|
Tumor tissue
|
[329]
|
|
CEP55
|
Cell cycle regulation, cytokinesis
|
↑ mRNA, protein
|
Tumor tissue
|
[112]
|
|
HSulf-1
|
Modulation of heparin-binding growth factor signaling
|
↓ mRNA
|
HNSCC cell lines
|
[330]
|
|
COL17A1
|
Cell interactions
|
↓ mRNA
|
Tumor tissue
|
[118]
|
|
Cortactin
|
Cell motility and invasion
|
↑ mRNA, protein
|
Tumor tissue
|
|
|
CXCR4
|
Chemokine receptor
|
↑ mRNA, protein
|
Tumor tissue, HNSCC cell lines
|
|
|
DAPK3
|
Apoptosis
|
↓ mRNA
|
Tumor tissue
|
[118]
|
|
DDR2
|
Receptor tyrosine kinase
|
↑ mRNA, protein
|
Tumor tissue, HNSCC cell lines
|
[337]
|
|
DSG3
|
Cell-to-cell adhesion
|
↑ protein
|
Tumor tissue, lymph nodes, HNSCC cell lines
|
[338]
|
|
DST
|
Regulation of the cell cycle
|
↓ mRNA
|
Tumor tissue
|
[118]
|
|
Hif-1α
|
TF, hypoxia
|
↑ protein
|
Tumor tissue, HNSCC cell lines
|
|
|
IL18
|
Apoptosis
|
↓ mRNA
|
Tumor tissue
|
[118]
|
|
LRP6
|
Signaling
|
↑ mRNA
|
Tumor tissue
|
[118]
|
|
Nuclear LKB1
|
Serine/threonine kinase
|
↓ nuclear protein
|
Tumor tissue
|
[340]
|
|
LOX
|
Lysyl oxidase family, important modulators of the ECM
|
↑ mRNA, protein
|
Tumor tissue
|
[341]
|
|
MMP-2
|
ECM degradation
|
↑ mRNA, protein
|
Serum, HNSCC cell line, tumor tissue
|
|
|
MMP-3
|
ECM degradation
|
↑ mRNA, protein
|
Serum
|
[344]
|
|
MMP-7
|
ECM degradation
|
↑ mRNA, protein
|
Tumor tissue, HNSCC cell lines
|
[55]
|
|
MMP-9
|
ECM degradation
|
↑ protein
|
Serum, tumor tissue
|
|
|
MYCN
|
Signaling
|
↑ mRNA
|
Tumor tissue
|
[118]
|
|
NBS1
|
Cell cycle regulation, DNA double-strand break repair
|
↑ mRNA, protein
|
Tumor tissue, HNSCC cell line
|
|
|
NF-κB
|
Proinflammatory TF
|
↑ mRNA, protein
|
Tumor tissue, HNSCC cell lines
|
|
|
Notch1
|
Single-pass transmembrane receptor
|
↑ protein
|
Tumor tissue
|
[349]
|
|
PPP2R1B
|
Apoptosis
|
↓ mRNA
|
Tumor tissue
|
[118]
|
|
RAC1
|
Rho family of Ras-related guanosine triphosphate (GTP)–binding proteins
|
↑ protein
|
HNSCC cell lines
|
[350]
|
|
RSK2
|
Cell cycle regulation, proliferation, apoptosis
|
↑ protein
|
Tumor tissue, HNSCC cell lines
|
[351]
|
|
Smad2
|
Mediates TGFβ signaling
|
↓ mRNA, protein
|
Tumor tissue
|
|
|
Snail
|
TF, regulator of EMT
|
↑ protein
|
Tumor tissue, HNSCC cell line, CSC-like subpopulation of tumor cells
|
|
|
Survivin
|
Inhibitor of apoptosis
|
↑ protein
|
Tumor tissue, HNSCC cell lines
|
|
|
Twist
|
TF, regulator of EMT
|
↑ protein
|
Tumor tissue
|
|
|
TLR-4
|
Interleukin-1R receptor family
|
↑ mRNA, protein
|
Tumor tissue, HNSCC cell lines
|
|
|
TRIM-29
|
TF
|
↑ mRNA, protein
|
Tumor tissue, HNSCC cell lines
|
[55]
|
|
VEGF/R
|
Angiogenesis
|
↑ protein, mRNA
|
Serum, tumor tissue, HNSCC cell lines
|
|
|
miR10b, miR211, miR181,
miR31
|
Gene expression regulators
|
↑
|
Plasma, HNSCC cell lines, tumor tissue
|
|
|
miR138,
miR34c,
miR203
miR200c
miR141
|
Gene expression regulators
|
↓
|
HNSCC cell lines, tumor tissue
|
7.9 Cell Death Modifications
Impairment of apoptosis is an important contributor to the development of HNSCC. HNSCC cells evolve many survival mechanisms that allow malignant cells to evade apoptosis. One of these mechanisms is loss of function of TP53 tumor suppressor. TP53 is a crucial damage sensor that triggers apoptosis [1]. Alternatively, tumors may evade apoptosis by elevated expression of anti-apoptotic agents (Bcl-2, Bcl-xL) [359] and survival signals (survivin [262, 354, 355]), by downregulation of proapoptotic factors (Bax, Bim, Puma) [360], or by mechanisms associated with cell cycle disruption. In particular, Bcl-xL and Bcl-2 overexpression strongly contribute to the development of HNSCC [361]; they inhibit apoptosis through prevention of cytochrome c release from mitochondria which impedes formation of the apoptosome, caspase activation, and cell death. NF-ĸB is also a major survival factor able to upregulate many survival proteins including Bcl-2 [134, 149, 323, 324, 348, 362]. Activation of the Wnt/β-catenin signaling pathway inhibits mitochondria-mediated apoptosis in HNSCC [363]. MiR-21 was found to target PTEN, programmed cell death 4 gene (PDCD4), and tropomyosin 1 (TPM1) to inhibit apoptosis, stimulate transformation, and enhance colony formation [364–366]. Recent studies demonstrated that miR-21 is also a key player in the STAT3 anti-apoptotic signaling pathway [147].
Anoikis is a type of programmed cell death that liquidates anchorage-dependent cells when they become detached from the extracellular matrix (ECM); however, the detached cancer cells are often resistant to chemotherapy or other cytotoxic and inflammatory stresses and do not undergo anoikis. Anoikis is caused by many factors including insufficient energy production. Osteopontin is able to upregulate energy metabolism. Interaction between osteopontin-a, which elevates the cellular glucose level, and osteopontin-c, which exploits this glucose to ensure energy production, is important to overcome conditions that lead to anoikis [367]. Resistance to anoikis is a critical step in metastasis as cancer cells are better able to survive during invasion and intrusion into the blood and lymphatic system [368]. Wnt-/β-catenin–mediated inhibition of apoptosis and anoikis is dependent on the death receptor signaling pathway [363]. Mutation of TP53 can also participate in resistance to anoikis. For example, serine substitution of proline at codon 151 of TP53 is a gain-of-function mutation leading to anoikis resistance and tumor progression in HNSCC [369]. Furthermore, growth factors secreted by endothelial cells (e.g., VEGF) could protect HNSCC against anoikis [370]. 14-3-3 zeta [252], Bcl-x(L) [361], Mcl-1 [371], CAV-1 [55, 325], and c-FLIP [329] also repress anoikis in HNSCC cells. Bit1 is downregulated in metastatic cancer [368]. Other well-known key players in HNSCC such as IGF-1R, EGFR, MAPK, RAS, NF-ĸB, Twist, RAC1, and TGF-β have been shown to suppress anoikis [372, 373].
Autophagy is an important physiological process that is usually very low during homeostasis that can be induced by cellular stress [374]. The induction of autophagy together with blocked apoptosis in hypoxic areas allows survival of tumor cells and promotes an aggressive phenotype in immunocompetent murine HNSCC models. On the other hand, autophagy defects are associated with neurodegeneration, liver failure, aging, and some types of cancer [375]. The role of autophagy in cancerogenesis is complicated and probably context-dependent. On one hand, failure to induce autophagy promotes cancer initiation due to persistent oxidative stress, DNA damage, and inflammation. On the other hand, a high level of autophagy facilitates survival of cancer cells under starvation and probably contributes to treatment resistance [376–378]. Schaaf et al. demonstrated that ULK1, unfolded protein response (UPR), and hypoxia-regulated genes are associated with autophagy induction, hypoxia tolerance, and a worse clinical outcome in HNSCC patients [379]. ULK1 triggers autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase [380].
7.10 Biomarkers of Treatment Response
Many biomarkers associated with drug resistance that are common in cancer stem cells, EMT, and resistance to cell death were discussed earlier. Chemical inhibition of the Ras-related guanosine triphosphate (GTP)–binding protein Rac1, which controls the organization of the actin cytoskeleton thereby regulating cell adhesion, polarity, motility, and EMT, resulted in significant improvement of HNSCC sensitivity to ionizing radiation and cisplatin [373]. Treatment-resistant HNSCC were reported to be CD44 high/EGFR low [381]. Furthermore, many conventional therapies fail because they utilize apoptotic mechanisms that require intact p53 signaling [382] or that can be inhibited by anti-apoptotic proteins such as Bcl-xL or Bcl-2 [383, 384]. Bcl-xL expression was correlated with resistance to radiotherapy (regardless of p53 status) [385, 386] and with resistance to chemotherapeutic agents such as cisplatin, bleomycin, vincristine, doxorubicin, and etoposide [385]. High Bcl-2 expression inhibits cisplatin-induced apoptosis and predicts poor response to cisplatin therapy in advanced OSCC [387]. In univariate and in multivariate analyses of tumor biopsy specimens, the simultaneous detection of bcl-2 protein overexpression and p53 gene mutation was associated with greater risk of locoregional failure and worse 5-year survival in HNSCC patients treated by radiotherapy [384]. Downregulation of miR-296 could significantly decrease the expression of Bcl-2, P-glycoprotein, and MDR1 and upregulate the expression of Bax [388].
Hypoxia in tumors modulates release of many angiogenic factors and cytokines and is associated with resistance to radiotherapy, treatment failure, and worse prognosis in HNSCC patients. Toustrup et al. developed a hypoxia classifier based on the expression of 15 genes [389]. The expression of miR-210 in head and neck cancer was in line with other approaches for assessing hypoxia and was correlated with locoregional disease recurrence and poor overall survival [390]. Hypoxia is also involved in creation of polyploid giant cancer cells that are important contributors to resistance [2, 391]. Moreover, the interaction between HIF-α/MIF and NF-ĸB/IL-6 axes plays an important role in the hypoxia-induced accumulation of MDSCs and tumor growth in HNSCC [392].
Excision repair cross complementation group 1 (ERCC1) is a crucial element in repairing of interstrand DNA cross-links. Accordingly, ERCC1 may cause resistance to mitomycin C (MMC) and platinum chemotherapeutics and might be used to predict a reduced therapeutic response [393, 394]. ERCC1+ cells exhibit elevated chemoresistance and appeared to be radiosensitive and less hypoxic [395]. High ERCC1 expression was associated with better overall survival rates in HNSCC. Nevertheless, patients with oropharynx SCC presenting with high ERCC1 expression remain disease-free and have increased survival rates when compared to non-oropharyngeal squamous cell carcinoma patients with high ERCC1 expression despite treatment modalities and HPV status [396, 397].
Oral cancer overexpressed 1 (ORAOV1) seems to be a crucial oncogene in OSCC. The ORAOV1 gene is often amplified in esophageal SCC and is reported to enhance tumorigenicity and tumor growth and, together with pyrroline-5-carboxylate reductase (PYCR), is associated with resistance to oxidative stress–generating therapies [398]. Moreover, the antioxidant capacity of lactate may also contribute to radioresistance in malignant tumors [399].
Subpopulations of Hoechst 33342 dye–resistant cells termed “side population” (SP) cells manifest stem cell characteristics [272]. Hoechst 33342 dye and also many kinds of cytostatics are exported out of the cell by the ATP-binding cassette (ABC) family of proteins including MRP1, MDR1, ABCG2, and ABCB5 [401, 402]. SP cells from OSCC are more chemoresistant, are more tumorigenic, and show self-renewal characteristics [272]. Finally, ABCC2/G2 inhibition in HNSCC cells with MK571 sodium salt hydrate markedly enhanced cisplatin sensitivity [400]. It was also suggested that the expression of ABCG2 is predictive for tumorous transformation of oral leukoplakia [401]. Likewise, elevated ABCB5 expression correlated with OSCC recurrence and progression [402]. ABCG2 and podoplanin protein expression may be useful markers of erythroplakia progression; 91 % of erythroplakic areas showing immunohistochemical positivity for both markers in the suprabasal layer of the oral epithelium became cancerous [403].
Cells in the tumor microenvironment (TME) have also been shown to induce tumor chemoresistance: (1) by the local release of cytokines that promote tumor growth and survival, (2) through specific interactions between tumor cells and ECM, (3) by oncologic trogocytosis, (4) by the transformation of primary cancer cells into cancer stem cells, and (5) by the creation of unique niches of tumor cells that provide survival advantages to the tumor following anticancer drug exposure or hypoxia (including upregulation of growth factors production by the stromal cells) [404].
7.11 Deregulation of Cellular Energetics
HNSCC cellular profiles have demonstrated many differences in the concentration of many metabolites, including lactate, fumarate, aspartate, alanine, phenylalanine, glycine, isoleucine, tyrosine, valine, taurine, NAD+, acetate, creatine, glutamine, UDP-sugars, myoinositol glutathione, and AMP/ADP [405]. As glucose is the main fuel required for ATP production in HNSCC cells [406], glutamine and glutaminolysis appear to be a major carbon source [405]. The glutamate/glutamine ratio was increased in HNSCC cells compared to normal human oral keratinocytes, indicating elevated glutaminolysis [405]. Glucose transporter GLUT1 expression was associated with poor survival and increased tumor growth [407].
Many in vitro metabolic studies have indicated that HNSCC cells are glycolytic with increased L-lactate production (Warburg effect). Recent in vivo studies demonstrated that metabolic diversity and metabolic symbiosis occur in HNSCC (see Fig. 7.4) [53]. The idea of a symbiotic relationship within the tumor microenvironment is supported by the fact that the Warburg effect exists in stromal cells rather than in cancer cells themselves. Cancer cells are able to consume lactate generated by stromal cells [408–411]. Accordingly, increased lactate levels in HNSCC tumors are associated with the occurrence of metastases [412, 413]. Curry et al. reported that MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in HNSCC [53]. MCT4 transports L-lactate and ketone bodies out of cells and may also serve as a biomarker of oxidative stress [414–416]. On the contrary, MCT1 transporter imports ketone bodies and L-lactate into the cell. High MCT1 expression causes an increase in oxidative phosphorylation (OXPHOS) and decreases glycolysis [411, 417]. The expression of MCT4 is triggered during hypoxia and oxidative stress through the activation of HIF-1α [418]. Significant MCT4 immunostaining was discovered in a vast majority of cancer-associated fibroblasts (CAFs) and differentiated carcinoma cells and was absent in adjacent normal stromal fibroblasts [53].
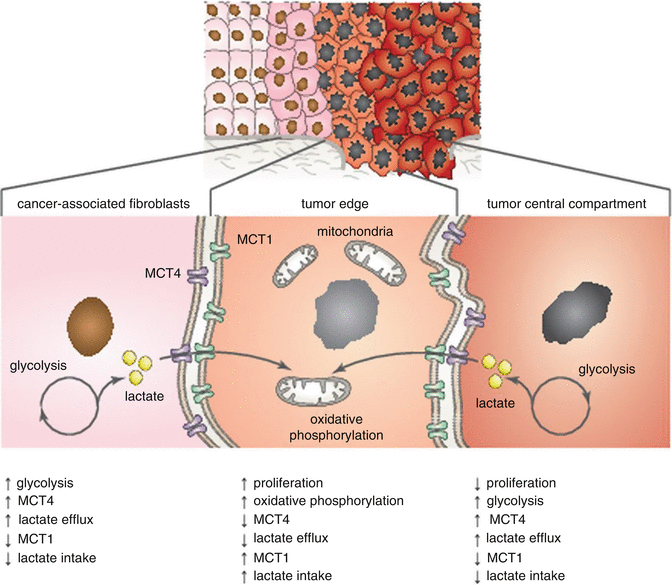
Fig. 7.4
Metabolic symbiosis in HNSCC tumor tissue. Highly proliferative cancer cells rely on oxidative phosphorylation and are mitochondrial-rich with high expression of MCT1. MCT1 transporter imports ketone bodies and L-lactate into the cell. Cancer-associated fibroblasts (CAFs) and nonproliferative cancer cells rely on glycolysis and are mitochondrial-poor with high expression of MCT4. MCT4 transports L-lactate and ketone bodies out of cells. Cancer cells are then able to consume lactate generated by stromal cells (Adapted from Curry et al. [53])
Interestingly, the cell proliferative index of cancer cells (determined by Ki-67 expression) was highly correlated with OXPHOS and MCT1 expression (p < 0.001). According to the metabolic characteristics, three distinct tumor areas were disclosed: (1) mitochondrial-rich and highly proliferative cancer cells (MCT1+/low MCT4/Ki-67+/TOMM20+/COX+), (2) mitochondrial-poor and nonproliferative cancer cells (MCT1−/MCT4+/Ki-67−/TOMM20−/COX−), and (3) mitochondrial-poor and nonproliferative stromal cells (MCT1-/MCT4+/Ki-67−/TOMM20-/COX-) [53]. Nonproliferative populations (Ki-67−/MCT4+) have also been shown to influence clinical outcome, possibly due to the ability to provide an energy source for highly proliferative subpopulations of cancer cells. MCT4 positivity in low-proliferative epithelial cancer cells was associated with poor clinical outcome (tumor recurrence; p < 0.0001). Similarly, MCT4 positivity was a specific marker for CAFs (p < 0.001) and was predictive for higher tumor stages (p < 0.03) [53].
7.12 Tumor Microenvironment
Tumors are not merely huddles of malignant cells but are in fact complicated well-organized complex tissue networks able to recruit many other cell types. Cooperation between tumor cells and nonmalignant cells creates the tumor microenvironment (TME). The nonmalignant TME cells play a complex role and promote all stages of tumorigenesis [1]. The TME in HNSCC includes many different cell types such as carcinoma-associated fibroblasts (CAFs), normal fibroblasts, myofibroblasts, endothelial cells (and their precursors), smooth muscle cells, pericytes, and immune cells [419]. Key TME cells and their properties are summarized in Table 7.5.
Table 7.5
Key cells of the HNSCC tissue microenvironment
|
Cell type
|
Markers
|
Secreted factors
|
References
|
|---|---|---|---|
|
Cancer-associated fibroblasts
|
α-SMA, integrin α6, FAP, tenascin-C, desmin, NG2, POSTN, PDGFR α/β, palladin, podoplanin, MCT4+, MCT1-
|
HGF, FGF-2,IGF-2, CCL-2, CXCL-1, CXCL-5, CXCL-12, TGF-β, MMP-2, MMP-9, VEGF, PDGF, type IV collagen, Col15-binding integrins, tenascin-C and tenascin-W, PGE-2
|
[440]
|
|
Regulatory T cells
|
CD4+CD25+FoxP3+
|
IL-10, IL-12, TGF-β
|
|
|
Cytotoxic T cells
|
CD8+, TCR, Fas, PD-1
|
Perforin, granzymes, granulysin
|
|
|
Th2 suppressor cells
|
CD4+
|
IL-4, IL-6, IL-10
|
|
|
CD34+ progenitor cells
|
CD34+
|
TGF-β
|
|
|
MDSCs
|
CD11b+ Gr1+ CD31+
|
IL-10, arginase, ROS, RNS, VEGF
|
|
|
Th17
T helper cells
|
CD4+, CD161 downregulation, CCR6, IL-23R, ROR2C
|
IL-17A, IL-17F, IL −21, IL-22, IL-26
|
|
|
Tumor-associated macrophages (M2)
|
IL-10, IL-6, PDGF TGF-β, MIF, EGF, CSF-1, MMP-9, CXCL-2,CXCL-8, VEGF, ROS, RNS, PGEs, CCL-17, CCL-18, CCL-22
|
||
|
Tumor-associated neutrophils
|
MMP-9, VEGF, HGF, elastase, ROS, PGEs
|
||
|
Endothelial cells
|
IL-6, CXCL1, CXCL8, endothelins
|
CAFs are critical elements of the TME participating in invasion, proliferation, and metastasis. These cells originate in normal fibroblasts located in close proximity to the tumor or in circulating mesenchymal stem cells [420]. Mutation of TP53 in tumor cells is associated with increased CAF migration to the TME, while intact p53 protein inhibits migration [421]. Oral CAFs exhibit cytokeratins, MMP-2, vimentin, and α-SMA–positive staining, whereas normal oral fibroblasts express only vimentin [159]. Marsh et al. highlighted the limited prognostic value of TNM staging and suggested that an SMA-positive stroma rich for myofibroblasts might be a powerful marker for the prediction of OSCC mortality [422]. CAFs express high levels of MCT4, which is a marker of glycolytic metabolism, oxidative stress, and L-lactate and ketone bodies export from the cell [53].
Proinflammatory factors known to be chemoattractants for macrophages and neutrophils are significantly upregulated in CAFs (e.g., CXCL-1, CXCL-5, and CCL-2) [423–425]. CAF-produced FGF-2 and VEGF then boost the process of angiogenesis. Known factors that stimulate tumor cell proliferation (SDF-1/CXCL-12, insulin-like growth factor-2 (IGF-2), and HGF) are also secreted by CAFs. Moreover, CAFs enhance tumor invasion through the secretion of tenascin-C, tenascin-W, HGF, and MMPs along with TGF-β [420, 426]. CAFs and cancer cells can produce sphingosine-1-phosphate (S1P) and prostaglandin E2 (generated by COX-2 activation) that can cause chemoresistance and can enhance cell survival through the PI3K-Akt/PKB pathway [427, 428]. Taken together, CAFs can promote tumor progression in several aspects through their ability to produce elevated levels of various growth factors, cytokines, ECM proteins, and MMPs [429].
The ability of cancer cells to produce angiogenic factors to create and maintain tumor vasculature has been well described; and the important influence of factors produced by endothelial cells on the tumor cells, in this regard, is also known. Gene expression analysis demonstrated that EGF, IL-6, and IL-8 are upregulated in cocultures of endothelial cells and HNSCC [430]. Inactivation of EGF, IL-6, or IL-8 secreted by endothelial cells (with antibodies or gene silencing) blocked phosphorylation of Akt, ERK, and STAT3 in tumor cells. Bcl-2 signaling is known to trigger the production of EGF, IL-6, and IL-8, which could be a mechanism for the upregulation of these cytokines in tumor-affected endothelial cells [430]. Bcl-2 gene expression is remarkably higher in the tumor-associated endothelial cells of HNSCC patients compared to endothelial cells derived from the normal oral mucosa [431]. Zeng et al. reported that Jagged1 (the Notch ligand), induced via growth factors and mitogen-activating protein kinase, caused Notch activation in endothelial cells adjacent to HNSCC and promoted capillary-like sprout formation [432]. Mast cells, also known as a mastocytes, are granulocytes that directly cooperate with endothelial cells to stimulate angiogenesis [433]. Enhanced mastocyte numbers during HNSCC progression correlated with angiogenesis [434, 435].
Macrophages are usually the most common immune cell population in the TME. Recruitment of circulating monocyte precursors triggers their differentiation into tumor-associated macrophages (TAM). The CCL-2 chemokine produced by tumor cells may be an important TAM chemoattractant along with other molecules such as TGF, VEGF, PDGF, and macrophage colony-stimulating factor (M-CSF) [134]. The phenotype of TAM resembles M2 cells with pro-tumor functions such as inhibition of Th1 adaptive immunity, production of growth and survival factors (e.g., EGF, IL-6, and IL-8), the secretion of angiogenic factors (e.g., VEGF, PDGF, TGF), and a variety of chemokines. TAM can also induce the degradation and remodeling of ECM, via the expression of MMPs, and suppress Th1 adaptive immune responses through the production of immunosuppressive mediators (e.g., IL-10, PGE2, TGF) and chemokines capable of recruiting Th2 cells (e.g., CCL-17, CCL-18, and CCL-22) [436, 437]. Taken together, TAM plays an important role in support of tissue remodeling, tumor progression, and angiogenesis, while suppressing Th1-type adaptive immunity [438].
Tumor cells are challenging opponents. The high level of genetic instability in cancer has proven to be a difficult barrier to overcome with specifically targeted therapies. Tremendous redundancy of the key pathways driving tumorigenesis is another challenge that we have not been able to conquer and, with single target therapies, probably never will. It is necessary to achieve complex knowledge of tumor biology in order to create more effective therapies. If we are not able to hit tumor cells directly, therapies might need to be focused to eliminate malignant collaborators or to interrupt communication between tumor cells and their host cells in the TME. CAFs appear to be genetically stable and might provide reliable targets for immunotherapy [439]. Activated CAFs express some specific molecules such as alpha-smooth muscle actin (α-SMA), fibroblast activated protein (FAP), tenascin-C (TN-C), periostin (POSTN), neuron-glial antigen2 (NG2), PDGFR α/β, podoplanin, and palladin [440]. TME-directed therapies can also target proteins and networks that mediate crosstalk within tumor stroma. For example, targeting Galectin-1 in CAFs inhibited OSCC metastasis by downregulating MCP-1/CCL2 expression [425]. A more comprehensive approach such as this, in which the complexity of malignancy is taken into account, could produce promising results and provide more effective treatments.
Acknowledgment
This work was supported by Ministry of Health of the Czech Republic IGA MZ NT14337-3/2013.
References
1.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi:10.1016/j.cell.2011.02.013.PubMed
2.
Erenpreisa J, Cragg MS. Three steps to the immortality of cancer cells: senescence, polyploidy and self-renewal. Cancer Cell Int. 2013;13:92. doi:10.1186/1475-2867-13-92.PubMedCentralPubMed
3.
Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24(17):2666–72. doi:10.1200/jco2005.04.8306.PubMed
4.
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–37. doi:10.1038/35052073.PubMed
5.
Pedrero JMG, Carracedo DG, Pinto CM, Zapatero AH, Rodrigo JP, Nieto CS, et al. Frequent genetic and biochemical alterations of the PI3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer. 2005;114(2):242–8. doi:10.1002/ijc.20711.PubMed
6.
Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45(4–5):324–34. doi:10.1016/j.oraloncology.2008.07.011.PubMedCentralPubMed
7.
Chang SE, Bhatia P, Johnson NW, Morgan PR, McCormick F, Young B, et al. RAS mutations in united-kingdom examples of oral malignancies are infrequent. Int J Cancer. 1991;48(3):409–12. doi:10.1002/ijc.2910480318.PubMed
8.
White RA, Malkoski SP, Wang XJ. TGF beta signaling in head and neck squamous cell carcinoma. Oncogene. 2010;29(40):5437–46. doi:10.1038/onc.2010.306.PubMedCentralPubMed
9.
Saranath D, Chang SE, Bhoite LT, Panchal RG, Kerr IB, Mehta AR, et al. High-frequency mutation in codons 12 and 61 of h-RAS oncogene in chewing tobacco-related human oral-carcinoma in India. Br J Cancer. 1991;63(4):573–8. doi:10.1038/bjc.1991.133.PubMedCentralPubMed
10.
Lu SL, Herrington H, Reh D, Weber S, Bornstein S, Wang D, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20(10):1331–42. doi:10.1101/gad.1413306.PubMedCentralPubMed
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


