Drug
Molecular target(s)
Mechanism of action
Phase
Clinicaltrials.gov identifier
Rigosertib
AKT, PI3K, PLK1
Multi-TKI
II
NCT01807546
Dasatinib
BCR/ABL, SRC, c-KIT, etc.
Multi-TKI
II
NCT01488318, NCT00882583
Capmatinib
c-MET
TKI
I/II
NCT02205398
Tivantinib
c-MET
TKI
II
NCT01696955
Palbociclib
CDK-4, CDK-6
Serine/threonine kinase inhibitor
II
NCT02101034
EGFR antisense DNA
EGFR
Block EGFR translation
I/II, II
NCT00903461, NCT01592721
Erlotinib
EGFR
TKI
II
NCT00573989, NCT01064479, NCT01316757, NCT01580449
Nimotuzumab
EGFR
mAb
II, III
NCT00702481, NCT01345084,
Afatinib
EGFR, HER2
Multi-TKI
II, III
NCT01415674, NCT01538381, NCT01732640, NCT01824823, NCT01783587, NCT01856478
Lapatinib
EGFR, HER2
Multi-TKI
II
NCT01044433, NCT01612351
Dacomitinib
EGFR, HER2, and HER4
Multi-TKI
II
NCT01449201
Poziotinib
EGFR, HER2, and HER4
Multi-TKI
II
NCT02216916
MEHD7945A
EGFR, HER3
mAb – dual targeting
II
NCT01577173
Debio 1143
IAP
Inducing apoptosis
II
NCT02022098
Cixutumumab
IGF-1R
mAb
II
NCT00957853
IL-12
IL-12R/NK cells
Human cytokine, immunomodulating
II
NCT01468896
Plasmid IL-12
IL-12R/NK cells
Human cytokine, immunomodulating
II
NCT02345330
Trametinib
MEK1/2
Threonine/tyrosine kinase inhibitor
II
NCT01553851
Everolimus
mTOR
mTOR inhibitor
I/II
NCT01283334, NCT01111058, NCT01133678
Rapamycin
mTOR
mTOR inhibitor
II
NCT01195922
Nivolumab
PD-1
mAb
I/II, III
NCT02327078, NCT02105636
Pembrolizumab
PD-1
mAb, immunomodulating
II, III
NCT02296684, NCT02289209, NCT02252042, NCT02255097, NCT02358031
MEDI4736
PD-L1
mAb, immunomodulating
II, III
NCT02207530, NCT02319044, NCT02369874
Buparlisib
PI3K
TKI
II
NCT01527877, NCT01816984
BYL719
PI3KCA
TKI
I/II, II
NCT02298595, NCT01602315, NCT02145312
BI 2536
PLK1
Serine/threonine kinase inhibitor
II
NCT00526149
VTX-2337
TLR-8/NK cells
Small molecule agonist
II
NCT01836029
Bevacizumab
VEGFA
mAb
III
NCT00588770
Axitinib
VEGFR, c-KIT, PDGFR
multi-TKI
II
NCT01469546
Vandetanib
VEGFR, EGFR, RET
multi-TKI
II
NCT01414426
Sorafenib
VEGFR, PDGFR, RAF
multi-TKI
I/II
NCT02035527
Pazopanib
VEGFR, PDGFR, FGFR, c-KIT, etc.
multi-TKI
II
NCT01377298
MK-1775
WEE-1
TKI
II
NCT02196168
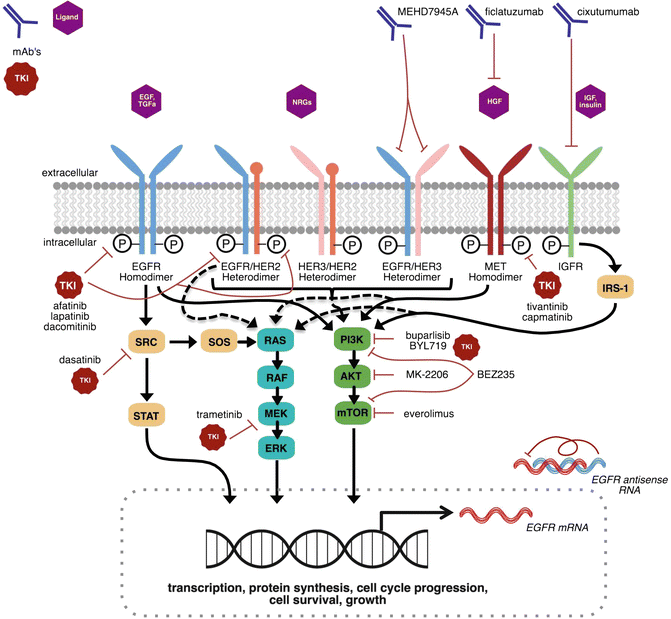
Fig. 8.1
Emerging molecular targets, including receptor tyrosine kinases and their downstream signaling pathways, for HNSCC treatment. EGFR family pathway signaling is enhanced or constitutively activated in HNSCC. Activation of alternative EGFR pathway RTKs (e.g., HER2, HER3) and non-EGFR RTKs (e.g., MET, IGFR) have been proposed as potential mechanisms of cetuximab resistance. Simultaneous targeting of multiple proteins with multiple drugs (i.e., EGFR and MET targeting using cetuximab and capmatinib, respectively) or using drugs with multiple targets (e.g., afatinib, lapatinib, dacomitinib) may delay resistance and improve survival. Downstream signaling converges through PI3K/AKT/mTOR, RAS/RAF/MEK/ERK, and SRC/STAT3 pathways to promote tumorigenic effects. Blocking downstream signaling through inhibition of these pathways is emerging as a viable strategy in select HNSCC populations. This figure illustrates the major emerging targets and their respective drugs in phase II/III clinical trials
8.2.1.1 EGFR
We and others previously reported that EGFR overexpression is an early event in HNSCC carcinogenesis where EGFR represents a plausible therapeutic target [44, 49]. Targeted therapies aimed at inhibiting EGFR include monoclonal antibodies (mAb) or tyrosine kinase inhibitors. mAbs prevent ligand-binding-induced receptor activation and induce EGFR internalization. Orally available anti-EGFR TKIs block EGFR kinase activity and uncouple downstream signaling. However, in randomized placebo-controlled phase III trials, EGFR-specific TKIs have shown 195 no benefit in the treatment of HNSCC [50, 51]. Lastly, antisense DNA inhibition EGFR, via intratumoral injection is an experimental approach to abrogate EGFR signaling at the pre-translational level [52]. Although EGFR is no longer an “emerging” therapeutic target, EGFR is central to, and prototypic of, strategies aimed at inhibiting aberrantly activated pathways in HNSCC and thus is discussed below.
Currently, EGFR is the only molecular target with a US Food and Drug Administration (FDA)-approved drug, cetuximab (Erbitux®), for the treatment of primary or R/M HNSCC [53]. First approved in 2006, cetuximab is a chimeric human/murine IgG1A monoclonal antibody (mAb) with high affinity toward EGFR. Cetuximab is approved for the treatment of R/M HNSCC in combination with radiation therapy, in combination platinum-based chemotherapy, and in combination with 5-fluorouracil (5-FU) for first-line treatment of R/M HNSCC after failure of platinum-based chemotherapy and thus currently is a standard of care. Cetuximab elicits its antitumor activities via inhibition of cell proliferation, activating antibody-dependent cell-mediated cytotoxicity (ADCC), as well as potentiating the cytotoxic effects of chemotherapy and radiation therapy [54, 55].
The utility of cetuximab, despite high initial promise, has been limited. Clinical response rates to cetuximab for the treatment of R/M HNSCC are approximately 13 % and extend survival by less than 3 months [19, 55]. Strategies to select HNSCC patients who will benefit from anti-EGFR therapy remain elusive and predictive biomarkers of cetuximab response have yet to be defined. Elevated EGFR protein expression is an indicator of poor prognosis and correlates with decreased overall survival (OS) and progression-free survival (PFS) [46–48, 56]. Likewise, EGFR gene copy number, nor EGFR protein expression, has been demonstrated to be predictive of cetuximab response in HNSCC [57]. Therefore, it suggests that in a background of high EGFR expression, cetuximab concentration may be suboptimal to block all receptors, thus maintaining downstream signaling. Additional EGFR mAbs (e.g., panitumumab, zalutumumab, nimotuzumab) are currently in various phases of preclinical and clinical evaluation [58]. Results of these studies have demonstrated that other EGFR mAb confer no additional benefit in patients with R/M HNSCC over cetuximab, although select agents may yield improved adverse reaction profiles [59–61]. EGFR-specific TKIs, although promising because they are orally bioavailable, have not proven effective in unselected HNSCC populations and randomized placebo-controlled phase III clinical trials have not demonstrated a clinical benefit [50, 51].
Despite promising clinical success of these agents, the majority of patients do not respond to anti-EGFR treatment and thus demonstrate intrinsic resistance to cetuximab therapy. In those patients initially responsive to treatment, virtually all eventually exhibit secondary or acquired resistance. Thus, overcoming resistance is paramount to extend the utility of these agents and improve outcomes in HNSCC patients. Potential mechanisms of anti-EGFR resistance include overexpression and heterodimerization of other ErbB family receptors, non-ErbB family RTK activation (e.g., IGF or MET), and constitutive activation of downstream effectors (e.g., RAS/RAF/MEK/ERK, PI3K/AKT/mTOR, and JAK/STAT3 pathways) [62–69]. Stimulation of these pathways may occur independent of EGFR pathway activation or through other stimuli such as hypoxia, induction of epithelial-mesenchymal transition (EMT), or activation of the angiogenesis pathways. Therefore, identification of biomarkers of EGFR-inhibitor response and druggable co-targets to overcome cetuximab and other EGFR-inhibitor resistance are critical.
8.2.1.2 EGFR mRNA
EGFR mRNA represents a plausible target in the class of EGFR-inhibiting therapies. EGFR antisense vectors, under the control of the U6 small nuclear RNA promoter, synthesize short antisense RNA sequences which complement sense EGFR mRNA transcripts (Fig. 8.1) [49]. In one preclinical study of intratumoral injection of EGFR antisense vectors, EGFR antisense therapy inhibited tumor growth, suppressed EGFR protein expression, and induced apoptosis in HNSCC xenografts [49]. In a subsequent series of experiments, combination treatment with docetaxel in HNSCC xenografts yielded synergistic effects on proliferation and inhibition of downstream signaling through MAPK, AKT, and STAT3 signaling pathways and extended the preclinical efficacy of cytotoxic chemotherapeutics [70]. A phase I safety and biological effect study of EGFR antisense therapy was completed in 17 advanced HNSCC patients who were refractory to standard therapies [52]. In this study, a maximum tolerable dose was not achieved, no severe adverse effects were reported, and 29 % and 12 % demonstrated clinical and complete responses, respectively [52]. Currently, two phase I/II clinical trials (NCT01592721 and NCT00903461) to investigate the efficacy of AS EGFR in combination with radiation and cetuximab are ongoing [58].
8.2.1.3 HER2
One proposed mechanism of resistance to EGFR-targeted therapy is sustained pathway signaling via alternate ErbB/HER family members [65, 71]. In addition to EGFR, there are three additional ErbB/HER RTKs: HER2/neu (ErbB2), HER3 (ErbB3), and HER4 (ErbB4) (Fig. 8.1) [32]. These ErbB/HER members can form heterodimers with EGFR or with other members including HER2/HER3, HER2/HER4, and HER3/HER4. These heterodimers transduce a variety of highly regulated cellular communiqué and provide redundant and complimentary signaling that promotes tumorigenesis and EGFR resistance.
ErbB2/HER2 was originally identified as the oncogene, neu, in mouse embryonic fibroblasts [72]. Subsequently, HER2 has been demonstrated to be overexpressed or amplified in a number of human cancers, including HNSCC [73]. Activation of HER2 elicits tumor-promoting effects including proliferation, cell motility, and survival in HNSCC cells [74, 75]. HER2, unlike the other ErbB receptors, lacks a ligand-binding domain, does not have a known ligand, and requires heterodimerization with other ErbB receptors for activity [76]. Alternatively, HER2 participates non-autonomously as a signal amplifier when in a heterodimerized state. Further, heterodimerization with HER2 facilitates increased receptor recycling to the cell membrane and delays ligand dissociation from ErbB family receptors [77, 78]. Studies of association between the ErbB family members implicate HER2 as the preferred binding partner of the other ErbB family members [77, 79, 80]. The specific downstream pathways stimulated by HER2 potentiation are dependent on ligand binding and heterodimerization partners [71]. EGFR ligand binding promotes the association of EGFR/HER2 heterodimers and preferentially activates the MAPK pathway [71]. Neuregulins (e.g., NRG1, NRG2), however, stimulate HER2/HER3 heterodimerization and activate both MAPK and PI3K/AKT pathways [71]. Thus, specific prolonged ligand binding and enhanced signaling, compounded by increased recycling to the cell membrane and preferential heterodimerization, supports HER2 targeting as a for the treatment of HNSCC.
We recently reviewed the role of HER2 in the molecular biology of HNSCC [71]. Rates of overexpression range from 0 to 47 % in HNSCC; however variability in detection techniques and reporting may complicate these findings [71, 81]. HER2 has been demonstrated to be overexpressed in a subset of HNSCC [73, 82–84]. We recently reported that 4 % of The Cancer Genome Atlas (TCGA) HNSCC tumors had increased mRNA expression, of which approximately half had HER2 amplification [71, 81]. HER2 overexpression correlates with poor prognosis, recurrence, and worse OS [73, 84, 85]. HER2 expression is higher in metastatic lymph nodes than the primary tumors and is predictive of advanced disease and nodal involvement [82]. However, a recent TCGA analysis found no association between either mRNA expression or increased total or phosphorylated HER2 and OS [71]. Alternatively, HER2 expression is correlated with EGFR expression and EGFR expression is an indicator of poor prognosis [71].
Co-expression of EGFR and HER2 is associated with cetuximab resistance in preclinical HNSCC models, and thus HER2 inhibition is a logical target to extend the utility of this class of drugs [62]. Given the central importance of EGFR and EGFR-targeted therapies in the management of HNSCC, and the activation of HER2 in a subset of HNSCCs, the majority of studies investigate HER2 inhibition in the context of EGFR-therapy resistance. As with EGFR-targeted therapies, there are two main strategies to inhibit HER2: mAbs and TKIs. Trastuzumab, a mAb with high specificity to HER2, is FDA approved for the treatment of HER2-positive carcinomas of the breast. In HNSCC cells, addition of trastuzumab to gefitinib yielded a synergistic effect on proliferation in 2 of 12 cell lines [5]. The utility of lapatinib in the treatment of HNSCC, a reversible dual EGFR/HER2 TKI, was recently reviewed by Gandhi and Agulnik [86]. Laptinib inhibits HNSCC cell growth and colony formation in vitro and in vivo and enhances the efficacy of platinum-based chemotherapeutics [87]. Other multi-ErbB family TKIs with HER2 activity in preclinical and clinical development include afatinib (EGFR/HER2) and dacomitinib (EGFR/HER2/HER4) [88]. Recent investigations using these inhibitors have focused on overcoming therapy resistance. In cetuximab-defiant HNSCC clones, resistance was enhanced by activation of HER family members and MET receptors [64]. Subsequently, resistance was abrogated by gefitinib and erlotinib and the experimental pan-HER TKI canertinib [64]. We demonstrated that the EGFR/HER2 targeting TKI, afatinib, overcame cetuximab resistance in HNSCC cells in vitro and in vivo and that co-treatment yielded supra-additive effects [62]. Dacomitinib inhibited the growth of the majority of HNSCC cell lines in vitro, including cetuximab-resistant lines, and blocked EGF-stimulated MAPK and PI3K/AKT/mTOR activation to a greater extent than cetuximab [89]. Dacomitinib treatment delayed tumor growth and demonstrated additive inhibition when combined with radiation in HNSCC xenografts [90].
Clinical trials evaluating HER2-targeting drugs have yielded mixed results. In a study investigating the effect of trastuzumab on response to chemotherapy in R/M HNSCC, no clinical responses were observed [91]. Thus, trastuzumab is no longer being evaluated in the treatment of HNSCC. Lapatinib, a reversible dual EGFR/HER2 TKI with US FDA approval for co-treatment with capecitabine in HER2-overexpressing metastatic carcinomas of the breast, has been evaluated in several phase II clinical trials in HNSCC [58]. Lapatinib, when combined with chemoradiation in advanced HNSCC, was associated with higher complete response (CR), PFS, and OS rates, and this effect was most profound in p16-negative tumors [92]. In another phase II trial, lapatinib, as a single agent in R/M HNSCC, failed to demonstrate effect on survival despite suppression of pHER2 expression [21]. A larger study of previously untreated locally advanced HNSCC, lapatinib alone for 2–6 weeks, followed by cisplatin-based chemoradiation, reported equivocal results between treatments [23]. The ORR was 17 % in patients receiving lapatinib alone for at least 4 weeks; however after chemoradiation, there were no significant differences [23]. Currently there is a single open-active phase II clinical trial evaluating lapatinib in combination with carboplatin and paclitaxel as induction therapy prior to transoral surgery and risk-adapted adjuvant therapy in HNSCC (NCT01612351) [58]. Afatinib is an irreversible dual EGFR/HER2 TKI that has FDA approval for the treatment of metastatic, EGFR-mutation-positive, non-small cell lung cancer. Afatinib has demonstrated activity in phase II clinical trials in R/M HNSCC with an ORR of 16 %, which was marginally better than cetuximab in a head-to-head trial (NCT00514943) [93]. Failure in one arm allowed crossover to the other arm, which highlighted potential activity in cetuximab-resistant tumors. Currently, there are eight open-active phase II/III clinical trials evaluating afatinib in HNSCC including maintenance therapy and neoadjuvant therapy with surgery, in combination with or after failure of chemotherapy, and in postsurgical patients at high risk of recurrence [58]. Similarly, dacomitinib is an irreversible EGFR/HER2/HER4 TKI in clinical development for the treatment of R/M HNSCC [94]. A recently completed phase II study in R/M HNSCC met its primary endpoint and demonstrated a 13 % ORR with median PFS of 12.1 weeks and had favorable tolerability [94]. A second phase II clinical trial in platinum-failed R/M HNSCC demonstrated a favorable toxicity profile with a 21 % ORR and 3.9 month PFS [95]. This study identified that activating PI3K mutations and the expression of pro-inflammatory genes were negative indicators of response to dacomitinib and might serve as biomarkers of response [95]. Another pan-HER TKI, poziotinib, is in phase II clinical trials (NCT02216916) in R/M HNSCC in patients refractory to platinum; however little preclinical data are available [37, 58]. HER2 remains a promising target in the treatment of HNSCC in the context of multi-EGFR targeting drugs.
8.2.1.4 HER3
The role of HER3 in EGFR pathway signaling, HNSCC tumorigenesis, and EGFR resistance is being defined. Unlike HER2, ErbB3/HER3 maintains a ligand-binding domain but lacks catalytic activity in the kinase domain [96]. HER3 has not been shown to homodimerize but it does preferentially heterodimerize with EGFR and HER2 to potentiate their kinase activity [53]. The cognate ligand for HER3 is neuregulin (NRG1/heregulin) which preferentially induces HER2/HER3 heterodimerization and phosphorylation of tyrosine residues in the kinase domain [53, 96]. These phosphorylated residues serve as an activation site for PI3K, a downstream target of EGFR pathway activation and potent oncogenic signaling mediator (Fig. 8.1) [53]. HER2/HER3 heterodimers elicit strong activation of the PI3K/AKT survival pathway emphasizing the importance of the HER3-PI3K pathway node in tumorigenesis [65]. It is proposed that HNSCC tumors require PI3K signaling to maintain a pro-survival phenotype [19, 55]. Therefore, blocking HER3 may inhibit pro-survival signaling via PI3K. However, because HER3 has intrinsically low levels of tyrosine kinase activity, the development of small molecule inhibitors has lagged.
Few studies have reported the prognostic significance of HER3 expression in HNSCC. Takikita et al. reported that membranous HER3 expression is associated with poor prognosis and decreased survival and was more predictive of decreased survival than HER2 expression [97]. In a study of 750 tumors, of which 127 were from the head and neck, expression of the HER3 cognate ligand NRG1 was highest in HNSCC in the absence of gene amplification. HER3 expression correlated with HER3 activation and was higher in metastatic lymph nodes [88, 98]. Oncogenic activation of HER3 is independent of activating genetic events such as amplification or mutation in HNSCC [88, 98]. Alternatively, oncogenic HER3 activation is driven in a NRG1-mediated autocrine feedback loop [98]. Thus it is likely that HER3 represents a driver of tumorigenesis and resistance to EGFR-targeted therapies in HNSCC and, like HER2, may be a candidate for molecular targeted therapies.
There are few preclinical studies investigating HER3-targeted therapies in HNSCC. The efficacy of HER3 mAb MM-121 was demonstrated in HNSCC models in vitro and in vivo, and additive effects were demonstrated when combined with cetuximab [99]. MEHD7945A, a dual-targeted mAb against EGFR and HER3, reported superior anticancer effects in HNSCC cells and xenografts and overcome acquired resistance to cetuximab or erlotinib treatment [66]. Furthermore, treatment with MEHD7945A overcame cross-resistance to radiation with enhanced DNA damage and abrogated pro-survival and DNA repair pathway signaling [66]. Currently, MEHD7945A is the only HER3-targeted therapy phase II for HNSCC (NCT01577173) [52, 58]. This open-label phase II clinical trial is comparing cetuximab therapy to MEHD7945A in patients who have progressed on platinum-based chemotherapies [58].
8.2.1.5 SRC
SRC is a cytoplasmic protein belonging to the Src-family of non-receptor tyrosine kinases (SFKs) including: YES, FYN, LYN, LCK, HCK, FGR, BLK, and YRK [57]. SRC integrates signal transduction from a range of stimuli including growth factors, integrins, and G protein-coupled receptors (Fig. 8.1) [100]. Aberrant SRC activation is implicated in tumorigenesis by stimulating proliferation, cell migration, invasion, epithelial-mesenchymal transition, angiogenesis, and metastasis [101, 102]. SRC is frequently activated in epithelial malignancies including HNSCC [100, 103]. In normal epithelium SRC expression is low and expression is progressively increased in inflamed, hyperplastic, and dysplastic oral epithelium and with advancement to squamous cell carcinoma [104]. In a Taiwanese cohort, high SRC protein expression was significantly associated with progression, recurrence, and poor prognosis [105]. Furthermore, aggressive clinical features (e.g., invasive phenotype, poor differentiation, and lymph node metastasis) correlate with activation of SRC in HNSCC [102]. In total, SFKs are activated (overexpressed) in up to 29 % of HNSCC with SRC specifically activated in 14 % of cases [106]. In addition, sustained SRC activation, through alternate receptor signaling pathways, provides an avenue of resistance to current RTK-targeted therapies [69, 107]. We reported that SRC is activated downstream of EGFR in HNSCC [108]. SRC activation mediates resistance to EGFR-targeted therapies such as cetuximab and TKIs, erlotinib, and gefitinib [63, 69, 109, 110]. Thus SRC represents a “node of convergence” and positions it as a promising target for therapeutic intervention [101, 107].
Preclinical evidence supports the continued investigation of SRC TKIs in the treatment of HNSCC. We demonstrated that SRC directly interacts with EGFR upon ligand binding, stimulating invasion, and proliferation of HNSCC cells, whereas SRC inhibition blocked EGFR-mediated proliferation and invasion [108, 111]. Likewise, saracatinib, a SFK-selective TKI, disrupts the invasive phenotype of HNSCC cells [112]. We found that saracatinib, when combined with gefitinib, abrogated gefitinib resistance with enhanced anti-tumor effects in HNSCC cells [110]. Dasatinib, an FDA-approved orally available multi-TKI, inhibited the proliferation, invasion, and cell migration and induced cell cycle arrest and apoptosis in HNSCC cells at clinically relevant concentrations [100]. We reported that combination treatment with erlotinib and dasatinib overcame ligand-independent (MET-mediated) erlotinib resistance and improved HNSCC inhibition in vitro and in vivo [69, 109]. It has also been shown that cetuximab resistance is mediated by SRC activation in HNSCC cells and that low-dose dasatinib could sensitize resistant cells [63]. Furthermore, dasatinib prevents radiation- and cetuximab-induced EGFR nuclear translocation and thus prevents EGFR-mediated DNA damage repair in vitro and in vivo [113–115]. In addition to MET, the insulin-like growth factor receptor (IGF1R) pathway is another RTK-activated pathway mediating EGFR-targeted therapy resistance [107]. Coadministration of dasatinib and an IGF1R TKI or mAb resulted in synergistic anticancer responses [107, 116]; and combined SRC and JAK2 inhibition resulted in a synergistic anti-tumor effect in patient-derived xenografts [117]. In summary, preclinical evidence suggests that SRC behaves like a hub for routing oncogenic signaling and resistance to pathway-targeted therapies.
Two SRC-targeting TKIs have been investigated in single-agent phase II clinical trials, saracatinib and dasatinib. Fury et al. evaluated saracatinib, a nonselective SFK TKI, in 9 patients with R/M HNSCC; however, the study was terminated early due to lack of activity (all patients had progressive disease) [118]. Because of its safety profile and poor clinical performance, the manufacturer of saracatinib is no longer supporting any continued clinical investigations [119]. Brooks et al. (2010) completed a phase II clinical trial investigating the efficacy of single-agent dasatinib, an orally available multi-TKI (i.e., BCR/ABL, SRC, c-KIT, ephrin receptors), in platinum-refractory R/M HNSCC and measured relevant cytokines and angiogenic factors [120]. This study failed to demonstrate significant activity as 12 of 15 patients evaluable for clinical responses demonstrated no ORs and only two (16 %) had stable disease (SD) despite a rapid and complete inhibition of SRC protein [120]. Based on the preclinical evidence, and the lack of efficacy of single-agent therapy, it is likely that the clinical utility of SRC may be in combination with targeted therapies such as cetuximab. Currently, only dasatinib is being investigated in two ongoing clinical trials in the treatment of HNSCC. The first is a phase I/II randomized clinical trial investigating dasatinib, cetuximab, and radiation, with or without cisplatin in locally advanced HNSCC (NCT00882583) [58]. The second is an open-label, single-arm phase II trial of cetuximab and dasatinib in cetuximab-resistant R/M HNSCC [58].
8.2.2 PI3K/AKT/mTOR Pathway
High-throughput sequencing of HNSCC tumors has elucidated a complex array of altered signaling pathways that converge on four distinct cellular processes: mitogenic signaling, differentiation, cell cycle, and apoptosis [121–123]. Mutated, aberrantly activated, and/or amplified genes within these pathways provide rational druggable targets for intervention. One of the major contributors to tumorigenesis in HNSCC is the activation of mitogenic signaling through the phosphoinositide 3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway [123]. PI3K/AKT/mTOR signaling controls critical cellular functions including survival, growth, and development and differentiation (Fig. 8.1) [71, 124]. Activation of EGFR family dimerization following ligand binding recruits PI3K family kinases to the membrane leading to the phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) and its conversion to the second messenger phosphatidylinositol (3,4,5)-trisphosphate (PIP3) [125]. PIP3 then serves as a membrane docking site for the serine/threonine protein kinase AKT (protein kinase B/PKB) [77, 78, 125]. AKT is then phosphorylated/activated by phosphoinositide-dependent kinase 1 (PDK1) and mTOR complex 2 (mTORC2). Conversely, PTEN negatively controls the activation of AKT by recycling PIP3 to PIP2. Activation of AKT has multiple downstream targets controlling cell survival, proliferation, angiogenesis, metabolism, and migration. A major downstream effector of pAKT is mTOR and the catalytic subunit mTOR complex 1 (mTORC1) [77, 79, 80, 125]. The kinase function of mTOR has two well-known substrates, S6 kinase 1 and 4E-BP1. Activation of S6K1 initiates protein synthesis whereas 4E-BP1 cancels the initiation of translation, thus antagonizing its action. Therefore, the major role of mTOR is the growth factor- and nutrient availability-dependent control of protein translation and likely the main mechanism of the anticancer effects elicited by rapamycin [71, 125]. Emerging druggable targets of this pathway include PIK3CA, AKT, and mTOR, and inhibitors are currently being evaluated in preclinical and clinical trials.
8.2.2.1 PI3K
Phosphatidylinositol-3-kinases (PI3Ks) are a family of signal transduction enzymes involved in basic cellular functions (e.g., cell growth, proliferation, differentiation, and survival) and are frequently implicated in cancers, including HNSCC [126]. To identify mutated, targetable pathways in HNSCC, we evaluated the mitogenic MAPK, JAK/STAT, and PI3K pathways using a whole-exome sequencing approach [81]. PI3K was the most commonly mutated oncogenic pathway and harbored mutations in 31 % of HNSCCs. JAK/STAT and MAPK pathways were mutated in 9 % and 8 % of tumors, respectively [81]. The most common gene mutated in HNSCC in this cohort was PIK3CA (13 %), which was also amplified in 24 % of tumors. Furthermore, in HPV-positive HNSCC, PI3K pathway mutations were the only oncogenic mutations identified in 20 % of cases. Other frequent PI3K pathway mutational events included PIK3CG and PTEN in 4 % of tumors. PI3K pathway-mutated tumors exhibited significantly more mutations than non-mutated tumors and enrichment of mutations in DNA damage/repair genes. Mutations in this pathway likely contribute to increased genomic instability and could potentiate tumorigenesis and resistance. Likewise, multiple mutations of PI3K pathway members are common (~22 %) in more advanced HNSCC. Rarely, HNSCC tumors harbored multiple mutations in MAPK (0 tumors) or JAK/STAT (1 tumor) pathway members.
Up to 90 % of mutations in PIK3CA gene are in the “hotspot” helical/kinase domains thus making the kinase activity of PIK3CA a potential therapeutic target [81]. Introduction of “hotspot” mutant PIK3CA constructs into HNSCC cells induced a significant growth advantage, enhanced PI3K pathway activation, and conferred sensitivity to mTOR/PI3K inhibitors in vitro [81]. Moreover, xenografts of “hotspot” mutant PIK3CA cell lines demonstrated that mutant PIK3CA were more sensitive to PI3K pathway inhibition than wild-type, independent of HPV status [81]. These findings suggest that activating PIK3CA mutations are potential biomarkers of response to PI3K pathway inhibitors. Furthermore, in tumor grafts harboring PIK3CA mutations, addition of mTOR/PI3K inhibitor to cetuximab treatment yielded additive effects [81]. These results were corroborated by Keysar et al. who evaluated PI3K and MAPK pathway activation in cetuximab-sensitive and cetuximab-resistant patient-derived xenografts (PDXs) demonstrated that PI3K pathway is activated in the setting of cetuximab resistance whereas MAPK pathway is dominant in cetuximab-sensitive cells [127]. Likewise, tumors harboring PIK3CA-activating mutations demonstrated enhanced PI3K pathway activation. An in vitro study of 64 HNSCC cell lines demonstrated that cell lines harboring PIK3CA mutations, but not amplification, were susceptible to PI3K inhibitors [1, 128]. PI3K inhibitor-resistant HNSCC cell lines demonstrated activation of the MEK/ERK pathway (pERK) after treatment with PI3K inhibitors [128]. Concomitant treatment with MEK and PI3K inhibitors yielded supra-additive inhibition and cell cycle arrest [128]. It is likely that HNSCCs depend on aberrantly activated PI3K signaling for tumor growth and represent a vulnerability that may be exploited with targeted therapeutics [123]. These results illustrate the potentially limited utility of PI3K inhibitor monotherapy in the background of multiple deregulated mitogenic pathways with significant crosstalk and emphasize the broader potential for co-targeted therapies and individualized medicine in the treatment of HNSCC.
Specific PI3K TKIs represent promising agents in the treatment of advanced and R/M HNSCC. Buparlisib (BKM120), a pan-isoform PI3K TKI, is one of the most advanced in clinical development and is currently being evaluated in early-phase trials as adjuvant therapy with paclitaxel (NCT01852292), cisplatin and radiotherapy (NCT02113878), and cetuximab (NCT01816984) [58]. In a recently completed phase Ib clinical trial in advanced solid tumors (NCT01283503) of two patients with HNSCC who were enrolled, one patient had progressive disease and the other had stable disease with an unconfirmed 76 % decrease in tumor volume [129]. BYL719 is αPIK3-specific TKI that is currently being evaluated in several clinical trials for HNSCC including: 1) in R/M HNSCC who have failed platinum-based chemotherapy (NCT02145312), 2) with cetuximab in R/M HNSCC who have failed platinum-based chemotherapy (NCT01602315), 3) with cetuximab and intensity-modulated radiation therapy (IMRT) in late-stage HNSCC (NCT02282371), and 4) with paclitaxel and cisplatin in HPV-associated oropharyngeal SCC (NCT02298595) [58]. The future utility of the PI3K pathway inhibitors continues to be defined. With personalized medicine on the horizon, the identification of patients who may glean benefit from PI3K inhibitors (e.g., those with PIK3CA activating mutations) may offer a more refined treatment strategy.
8.2.2.2 AKT
AKT is a group of three (AKT1-3) protein kinases downstream of PI3K that regulate normal cellular processes [e.g., cell survival (AKT1), and metabolism (AKT2)] as described above. Activation of AKT is frequently described in HNSCC and is a biomarker of poor prognosis [130–132]. The relationship of AKT to PI3K in transducing signaling implicates it as a possible molecular target in HNSCC [132, 133]. Mutations and amplifications in AKT are rarely reported in HNSCC [81, 134, 135]. However, constitutive activation of AKT may be maintained by upstream activation, PTEN deletion/mutation, and/or enhanced degradation of PTEN protein [125, 130, 131]. In addition to upstream activation by PI3K and/or loss of PTEN, AKT can also be activated in hypoxic conditions and serves as an independent prognostic indicator of radiosensitivity in HNSCC [130, 132, 136]. These considerations were highlighted in a clinical trial evaluating chemotherapy with cetuximab that demonstrated low PTEN expression was associated with reduced OS [137].
Clinical studies examining inhibition of AKT via targeted therapies are limited. A phase II study (NCT00062387) of perifosine, an alkylphospholipid with dual AKT and PI3K inhibitor effects, failed to demonstrate any response in R/M HNSCC and was terminated early [138]. More recently, in a comprehensive in vitro study of the efficacy of PI3K, mTOR, and an AKT inhibitors in HNSCC cell lines, the experimental AKT inhibitor (GSK690693) demonstrated efficacy across the most cell lines but did not decrease pAKT levels [128]. Another AKT inhibitor, MK-2206, inhibited HNSCC growth, migration, and metabolic activity in vitro [139]. Additionally, MK-2206 reduced xenograft tumor size, prevented cervical lymph node metastases, and improved survival in vivo [139]. The only specific AKT inhibitor being investigated in advanced R/M HNSCC and in clinical phases of development is MK-2206 (NCT01349933) [58]. This phase II clinical trial evaluated OS and PFS, in nasopharyngeal squamous cell carcinoma, although results have not yet been reported [58].
8.2.2.3 mTOR
Mammalian target of rapamycin (mTOR) was discovered as the eukaryotic molecular target of a soil-derived macrolide antibiotic, rapamycin. mTOR is a serine/threonine kinase which integrates cellular signals, including growth factors, nutrients, energy, and stress, to regulate protein synthesis, cell growth, and proliferation [140]. Cellular signaling via mTOR is achieved through two mTOR containing complexes, mTORC1 and mTORC2 [141]. mTORC1 is rapamycin-sensitive and comprised of mTOR, Raptor, and PRAS40. mTORC1 phosphorylates S6K1 and 4E-BP1 and is involved with protein translation and proliferation [142]. mTORC2, comprised of Rictor and Proctor, phosphorylates AKT and potentiates its enzymatic activity [142]. mTORC1 is tightly regulated by both the PI3K-AKT pathway and MAPK signaling pathways via tuberous sclerosis complex 2 (TSC2) [142]. Moreover, AKT/mTOR pathway activation is seen in several human malignancies, including 90 % of HNSCCs [143]. In HNSCC, mTOR activation can be signaled through EGFR-dependent or EGFR-independent mechanisms and represents an actionable molecular target for therapy [143].
mTOR specific “rapalogs” include rapamycin, temsirolimus, everolimus, and ridaforolimus. Rapamycin, the most comprehensively studied rapalog, has demonstrated profound anticancer effects in preclinical studies in vitro and in vivo [144–147]. In HNSCC xenografts rapamycin treatment decreased tumor size, blocked DNA synthesis, induced apoptosis, and reduced S6K1 activity [144]. Treatment with rapamycin or everolimus prevented intratumoral lymphangiogenesis and metastasis in a mouse orthotopic model of HNSCC [147]. Using a large collection of 52 patient-derived xenografts, treatment with everolimus had the third highest tumor response rate next to cetuximab and docetaxel [148]. Temsirolimus inhibited HNSCC xenograft tumor growth, and long-term administration prevented the outgrowth of minimal residual disease [149]. mTOR inhibitors may be useful in extending the utility of established HNSCC adjuvant treatment strategies. mTOR inhibition increases the radiosensitivity of head and neck cancer models [146]. Rapamycin, when combined with cetuximab or bevacizumab, and radiation, yielded enhanced anticancer effects in HNSCC xenografts [150]. Because of the robust preclinical data for mTOR inhibitors, continued clinical evaluation of this class of drugs is ongoing.
Rapalogs are in various phases of clinical development with everolimus being the most advanced in HNSCC. Fury et al. reported a phase I dose-escalation trial evaluating everolimus plus cisplatin in advanced solid tumors, of which nine were HNSCC [151]. Efficacy of everolimus treatment was reported in four patients who had been extensively pretreated with chemotherapy and radiation; however, all four regressed after 4–8 cycles [151]. In a follow-up to their first phase I trial involving everolimus, Fury et al. (2012) reported the results of another phase I trial examining everolimus plus cisplatin and intensity-modulated radiation (IMRT) in HNSCC patients [152]. In this small, preliminary study, the cohort had a PFS of 85 % and OS of 92 % with median follow-up of 19.4 months [152]. Fury et al. (2012) also reported a phase I study of temsirolimus plus carboplatin and paclitaxel and reported similar efficacy to their other studies [153]. However, a phase II study examining co-targeting of mTOR and EGFR using temsirolimus with erlotinib in R/M HNSCC was terminated early due to toxicity concerns [58]. Currently, there are three open studies examining everolimus (NCT01111058, NCT01133678) or rapamycin (NCT01195922) in phase I/II clinical trials [58].
The efficacy of single-agent, single-target rapalogs in the treatment of HNSCC is unlikely given a PI3K-dependent feedback loop via oncogenic activation of ERK1/2-MAPK pathway, which has been demonstrated in human clinical trials and verified in vitro [141]. The future utility of mTOR inhibition will likely utilize multi-targeted agents or “add-on” therapies to simultaneously block parallel signaling pathways to increase efficacy and prevent resistance [88, 154]. Dual targeting PI3K/mTOR and co-targeting mTOR and EGFR are being investigated in clinical trials. Co-targeting mTOR and EGFR, using everolimus plus cetuximab plus carboplatin in R/M HNSCC in a phase I clinical trial, yielded promising results where 62 % of participants demonstrated PRs with a median PFS of 8.2 months [155]. Treatment response was correlated with activated MAPK and mTOR, indicating a potential biomarker of response and illuminating the concept of personalized medicine [155]. The dual PI3K/mTOR inhibitor, BEZ235, was studied in advanced solid tumors, although no data are available for review [58, 128]. Currently, a phase I/II clinical trial evaluating the combination of BEZ235 and everolimus in advanced, unresectable tumors is underway (NCT01508104) [58]. However, it is ominous that a majority of clinical trials investigating BEZ235 have been terminated or withdrawn [58]. Although the utility of PI3K inhibitors as monotherapy may be limited, these data support the continued investigation of PI3K inhibitors to extend the efficacy of EGFR-targeted therapies, especially in defined patient cohorts.
8.2.3 MAPK Pathway: MEK1/2
8.2.3.1 MEK1/2
The mitogen-activated protein kinase (MAPK) pathway plays a central role in normal cellular function and is aberrantly activated in many human cancers including HNSCC [156, 157]. Interrogating targets that block activation of MAPK are a high priority for the treatment of HNSCC. One promising target is MEK, the canonical activator of ERK (Fig. 8.1) [158, 159]. The MAPK family of kinases include extracellular regulated kinases (ERK), cJUN terminal kinase (JNK), and p38 MAPK [116, 160]. The most frequently activated MAPK members signal through ERK and are associated with proliferation and survival [160]. Briefly, activation of the MAPK pathway occurs via RTKs such as EGFR family, mesenchymal-epithelial transition (MET) factor, insulin-like growth factor receptor (IGF1R), and fibroblast growth factor receptors (FGFR) [161–163]. Subsequent to growth factor ligand binding, RTKs undergo dimerization, transphosphorylation, and activation. Adaptor proteins (e.g., SHC, SOS, GRB) associate with activated RTKs and recruit guanine exchange factors (GEF) at the cell membrane. GEFs activate RAS GTPases (H-, N-, and KRAS) and transduce signals downstream through MAPK and PI3K/AKT/mTOR pathways [163]. Activation of the MAPK pathway is initiated by RAS-RAF kinase complex formation at the cell membrane and subsequent phosphorylation of RAF kinase. RAF kinase dissociates from inhibitor proteins and homo- or heterodimerizes to elicit MAPK kinase kinase (MAPKKK) activity. MAPKKK directly phosphorylates the dual-specific tyrosine and serine/threonine kinase MEK1/MEK2 and activated MEK is the sole activator of MAPK (ERK1/ERK2). Downstream targets of ERK activation include cell cycle proteins (e.g., cyclin D1) and transcription factors (e.g., MYC) that drive proliferation and survival. Additional downstream targets of ERK include SPRY and DUSP provide a negative feedback loop for MAPK pathway activation. Because ERK is the only known phosphorylation target of MEK it highlights a potential susceptibility in aberrant MAPK pathway activation. Currently, several MEK1/MEK2 inhibitors are being evaluated in early clinical studies to treat various cancers, including HNSCC.
In HNSCC, the oncogenic effects of MAPK pathway activation occur via RTK activation or through activating mutations in MAPK pathway members. Mutations in MAPK pathway are found in 8 % of tumors [164]. ERK is activated in up to 33 % of tumors [160]. Among the activating mutations in RAS, approximately 5 % hold HRAS mutations, but NRAS or KRAS mutations are rare [58, 121]. In HNSCC cells, MEK activation is directly associated with cetuximab and radioresistance [165, 166]. The experimental MEK1/MEK2 inhibitor, U0126, inhibited growth and potentiated the effect of radiotherapy in HNSCC cells [165]. MEK activation also induced resistance to PI3K inhibitors [128]. Co-treatment of HNSCC cells with an experimental MEK inhibitor, MEK 162, reestablished sensitivity to PI3K pathway inhibition and yielded additive and synergistic effects. Furthermore, these effects were more dramatic in HRAS mutant cells [128]. In the context of personalized medicine, in a subset of HNSCC tumors with HRAS mutations, MEK inhibition, alone or in combination with PI3K inhibitors, may be a viable treatment strategy.
MEK inhibitors are in various phases of clinical development for the treatment of a variety of solid and hematologic malignancies. The most advanced MEK inhibitor is trametinib. Trametinib was FDA approved in 2013 for the treatment of BRAF V600E-mutant melanoma [167]. In 2014, trametinib, in combination with BRAF inhibitor, dabrafenib, was FDA approved for the treatment of patients with BRAF V600E/K-mutant metastatic melanoma [167, 168]. A recently completed, but not yet published, phase I/II study examined trametinib with an experimental AKT inhibitor in multiple myeloma and solid tumors, including HNSCC (NCT01476137) [58]. A recently published, phase Ib clinical trial examined trametinib with mTOR inhibitor everolimus in advanced solid tumors [169]. Although two HNSCC patients were enrolled in this study, patient-level data for these cases were not available [169]. Currently, single-agent trametinib is being examined in a phase II window of opportunity trial in surgically resectable HNSCC (NCT01553851) [58].
8.2.4 Non-EGFR Pathway Receptor Tyrosine Kinases
8.2.4.1 MET Pathway
Like EGFR, mesenchymal-epithelial transition factor (MET) is a transmembrane RTK typically expressed on epithelial cells. Hepatocyte growth factor (HGF, aka scatter factor), secreted by mesenchymal cells and sometimes tumor cells, is the only known MET ligand. Upon ligand binding MET homodimerizes and autophosphorylates propagating signaling through PI3K/AKT/mTOR and RAS/RAF/ERK pathways (Fig. 8.1). Alternatively, receptor-type protein tyrosine phosphatase β (RPTP-β) negatively regulates MET activation through dephosphorylation [170]. In malignantly transformed tissues, MET is constitutively activated driving an aggressive cellular phenotype. In turn, this contributes to increased proliferation, angiogenesis, invasion, and metastasis in numerous cancers including HNSCC [171, 172]. Given its extensive involvement in oncogenesis, MET and its cognate ligand HGF are promising therapeutic targets in HNSCC.
MET expression in normal epithelium is low; however, its expression rises with progression from hyperplasia through increasing grades of dysplasia [173]. We and others have reported that MET is overexpressed in the vast majority of HNSCC (50–83 %); however, its prognostic significance has been debated [134, 174–178]. Activated (phosphorylated) MET has also been reported in the majority of HNSCC [130]. Several studies have demonstrated that MET expression is significantly higher in HNSCC with neck and lymph node metastases and increased depth of tumor invasion [172, 175, 179, 180]. Kim et al. reported that MET overexpression is associated with decreased survival in HNSCC of the oral tongue [176]. MET oncogene amplification and activating mutations are seen in a subset of HNSCC; however, in most cases the prognostic significance is unknown [181]. Interestingly, specific mutations (i.e., Y1235D) have been associated with a greater risk of distant metastasis in HNSCC [182]. Activation of the MET pathway supports invasive growth by stimulating epithelial-mesenchymal transition (EMT), a more stem-cell-like phenotype, degradation/transmigration of the basement membrane, migration through the extracellular matrix, and independence from apoptosis and anoikis (anchorage-dependent cell death) [172, 183]. HGF activation of MET positively regulates the expression of pro-angiogenic factors through MEK- and PI3K-dependent pathways [171, 184]. Overall, high MET expression correlates with worse survival and a more aggressive malignant phenotype.
Hepatocyte growth factor (HGF) is a plasminogen-related growth factor associated with epithelial remodeling and cell migration [185]. HGF is also the cognate ligand for the MET; thus, downstream cellular and oncogenic effects are communicated solely through MET [185, 186]. HGF overexpression is reported in several human malignancies including HNSCC, breast, colorectal, lung, cervical, hepatocellular, and others [186]. We demonstrated that 82 % of HNSCC overexpressed HGF when compared with matched adjacent normal mucosa [174]. Kwon et al. demonstrated that 100 % of HPV-negative tonsillar HNSCC expressed HGF and expression was associated with decreased OS and PFS and presence of lymph node metastasis [181]. Serum HGF correlates with initial stage, treatment (i.e., serum HGF is lower after chemotherapy or surgery), and prognosis (i.e., higher serum HGF is predictive of progression, recurrence, and diminished survival) in HNSCC [135, 176, 187]. Stromal HGF suppresses dendritic cell maturation and permits tumor immune escape [188]. Therefore, successful sequestration of HGF may block oncogenic MET signaling, serve as a peripheral blood biomarker for treatment efficacy, and even resensitize the host immune response to tumor.
Experimental inhibition of MET pathway can be accomplished through either direct inhibition of MET (via TKI or mAb) or sequestration of HGF with HGF-targeting mAbs. Met inhibition using small molecule inhibitors (crizotinib and SU1127) or siRNA in HNSCC in vitro abrogated MET expression and activation and reduced cell viability and migration [189]. In HNSCC xenograft studies, crizotinib treatment decreased proliferation and induced apoptosis and reduced tumor size [174, 190]. Knockdown of MET using siRNA in human HNSCC xenografts decreased proliferation and tumor growth, induced apoptosis, decreased lymph node metastasis, and increased survival [189]. When combined with EGFR inhibitor, erlotinib, or the chemotherapeutic, cisplatin, synergistic inhibitory effects on viability were demonstrated [69]. Dual inhibition of EGFR and MET using gefitinib and crizotinib reduced proliferation, invasion, and experimental wound healing greater than each alone and maximally suppressed downstream signaling through PI3K/AKT/mTOR and MAPK pathways [190]. Furthermore, we demonstrated that MET can be activated independently of HGF via TGFα-mediated activation of EGFR transduced activation of SRC [69, 190]. Unlike short-lived HGF-mediated activation of MET, EGFR-mediated activation was prolonged. Furthermore, knockdown of MET sensitized cells to EGFR TKI, erlotinib, in vitro and in vivo [69]. Therefore, we propose that EGFR and MET can compensate for one another through activation of shared signaling molecules (e.g., c-Src, Grb2, Ras, AKT, MAPK). These data support the notion that one mechanism of HNSCC resistance to EGFR-targeted therapies involves MET activation (and other RTKS) [186].
Unlike MET inhibitors, HGF-targeting mAbs (e.g., ficlatuzumab, rilotumumab, and AMG 102) are more nascent and preclinical data are sparsely available. We demonstrated that HGF is primarily expressed by mesenchymal cells and elicits its effects through paracrine oncogenic signaling in HNSCC cells [174]. Accordingly, enhanced invasion provoked by tumor-associated fibroblasts (TAFs), or stimulation with exogenous HGF, was abolished after treatment of HNSCC cells with a neutralizing HGF-targeting mAb targeting in vitro [174]. HGF activation of HNSCC cells stimulates a stem-cell-like cellular morphology and gene expression pattern [183]. However, robust preclinical studies examining HGF-targeted therapies in vivo have not been completed.
Several MET-targeting agents are in various phases of clinical development for the treatment of HNSCC. A single MET-targeting phase II clinical trial (NCT00725764) for the treatment of HNSCC has been completed utilizing foretinib, a multi-kinase inhibitor, with predominant activity toward MET and VEGFR2 [191]. Although this phase II trial failed to meet its primary endpoint, 50 % of patients maintained stable disease and 43 % demonstrated tumor shrinkage [191]. Another nonspecific MET multi-kinase inhibitor (with potent activity against MST1R, FLT3, AXL, MERTK, TEK, ROS1, DDR1/2, and MKNK1/2) LY2801653 is being examined in a phase I clinical trial (NCT01285037) in advanced solid tumors including a specific focus on a cohort of HNSCC tumors co-treated with cetuximab [58, 192]. Currently there are two active phase II clinical trials utilizing MET-specific TKIs [58]. The first examines the effectiveness of cetuximab with or without tivantinib in non-resectable or R/M HNSCC (NCT01696955). The second examines the effectiveness of combined capmantinib and cetuximab in MET-positive HNSCC after progression using cetuximab or panitumumab (NCT02205398) [58]. In addition to direct targeting of MET, abrogation of MET pathway signaling can be accomplished through mAb neutralization of its cognate ligand HGF. Currently, two HGF-targeting phase I trials evaluating the mAb ficlatuzumab are underway for R/M HNSCC in combination with cetuximab or with cisplatin and radiotherapy [58]. Overall, it is clear that targeting the MET pathway will remain the center of further investigations in treatment of HNSCC. Like other targets, MET pathway inhibitors may confer additional therapeutic benefit when used as add-on therapies aimed at overcoming cetuximab resistance. These therapies are emerging as a next line of strategies to improve outcomes in HNSCC and might identify specific subgroups of patients (MET activated) who may benefit.
8.2.4.2 IGF1R
Considerable evidence implicates insulin-like growth receptor 1R (IGF1R) in the development of epithelial cancers including HNSCC [193]. IGF1R belongs to the insulin receptor (IR) family of RTKs. With nearly ubiquitous expression in human cancers, IGF1R is an apparent candidate for targeted therapeutics [194]. Insulin, IGF1, and IGF2 are the known IGF1R ligands. Ligand binding induces autophosphorylation and activation of IGF1R [195]. The downstream target of activated IGF1R is insulin receptor substrate protein (IRS1) that initiates signaling through the PI3K/AKT/mTOR and MAPK pathways [195]. Activation of IGF1R regulates normal cellular functions such as cell cycle, differentiation, and motility [195]. In cancer IGF1R mediates the oncogenic signaling properties of IGF1, IGF2, and insulin, enhancing proliferation and survival and offering protection from apoptosis and anoikis [196]. IGFR1 and EGFR can interact directly through heterodimerization or indirectly through activation of overlapping signaling pathways [197]. This crosstalk may provide an avenue of resistance in malignant cells. In HNSCC, acquired resistance to EGFR-targeted therapy frequently implicates oncogenic signaling through IGF1R [197].
IGF1R is overexpressed in the vast majority of epithelial cancers including HNSCC. IGF1R is expressed in 77–100 % of HNSCC [193, 198, 199]. In one study, higher expression was associated with worse prognostic factors including local disease control (LDC), disease-free survival (DFS), and overall survival (OS) [199]. After controlling for stage, high IGF1R protein expression was predictive of worse prognosis in high-stage (III/IV) disease [199]. Others have reported high IGF1R expression in oral and oropharyngeal SCCs; however, expression was not predictive of poor prognosis (although they failed to control for HPV status, that in our opinion confounds their results) [198]. Additionally, levels of serum IGF1 are associated with increased risk of developing a second primary tumor in HNSCC patients [200].
Co-targeting constituents of adaptive cellular responses, including RKTs, may thwart targeted monotherapy resistance and augment anticancer effects in HNSCC. In a clinical trial examining efficacy, gefitinib failed to improve DFS, and shorter DFS was significantly associated with IGF1R overexpression [201]. In EGFR-inhibitor (gefitinib)-resistant cells, IGF1R and IRS-1 were constitutively activated sustaining PI3K/AKT/mTOR signaling [154]. These cells also demonstrated reduced expression of regulatory insulin growth factor binding proteins (IGFBP3/4) [154]. Co-targeting IGF1R and EGFR ameliorated gefitinib resistance [154]. Experimental activation of IGF1R partially reversed the cell cycle arrest caused by gefitinib in HNSCC cells [202]. IGF1R phosphorylation is also higher in PI3K inhibitor-resistant HNSCC cell lines and co-targeting IGF1R reverses the resistance [128]. Given that resistance to EGFR may involve multiple RTKs, including other HER family RTKs, Axelrod et al. recently demonstrated that concurrently targeting IGF1R, using an experimental TKI and a pan-HER-family TKI or SRC-family TKI, led to synergistic antiproliferative and pro-apoptotic effects in HNSCC cell lines [107]. In IGF1R mAb, (cixutumumab)-resistant HNSCC cells and xenograft inhibition of IGF1R with the mAb cixutumumab resulted in reciprocal activation of PI3K/AKT/mTOR pathway via activation of EGFR, AKT, and BIRC5 (survivin) [156]. Co-targeting mTOR (rapamycin) or EGFR (cetuximab) with IGF1R inhibition abrogated compensatory EGFR pathway signaling and synergistically enhanced antiproliferative and pro-apoptotic effects in HNSCC cells and xenografts [156]. Additionally, co-targeting EGFR and IGF1R enhanced sensitivity to radiation in HNSCC xenografts [158]. Therefore, in the context of EGFR pathway inhibition and resultant resistance, IGF1R targeting is another viable strategy to improve the utility of cetuximab or other EGFR-targeting drugs to improve outcomes in HNSCC.
Overall, clinical trials of anti-IGF1R mAbs, primarily in non-HNSCC patients, have demonstrated limited clinical efficacy in unselected patient populations [116, 203–206]. Likewise, in a phase II clinical trial, figitumumab monotherapy, an anti-IGF1R mAb, yielded no clinical benefit in patients with palliative HNSCC who had progressed after platinum-based therapy [58, 207]. However, a case report in a patient with recurrent inoperable cetuximab-resistant HNSCC purported that IGF1R inhibition may sensitize HNSCC patients to cytotoxic agents or that re-challenging with cetuximab might extend survival [208]. Although few conclusions can be drawn from this study, it provides valuable prospective molecular data on the oncogenic shift after treatment with different targeted therapies over time. A phase II study evaluating the combination of cetuximab and OSI-906, a dual TKI of IGF1R and IR, in HNSCC patients was recently withdrawn prior to enrollment after its parent company (Astellas Pharma, Inc.) shuttered operations of OSI Pharmaceuticals, Inc in 2013 [58, 209]. Another phase II trial (NCT01465815) evaluating erlotinib and OSI-906 in cutaneous HNSCC reported that the safety of the combination was unfavorable [58, 209]. Although compelling preclinical data for OSI-906 have been reported, it is likely that the safety profile is unfavorable and continued oncologic investigations in HNSCC are unlikely. A phase II trial (NCT00617734) evaluating the anti-IGF1R mAb cixutumumab (IMC-A12) as a single agent or in combination with cetuximab for R/M HNSCC was recently completed; the results are not yet published [58]. A second phase II clinical trial sponsored by the same group that is currently recruiting is evaluating biomarker modulation using preoperative cetuximab with or without cixutumumab in patients with surgically eligible HNSCC (NCT00957853) [58].
8.3 Cell Cycle Regulatory Targets
Proliferation of eukaryotic cells involves the well-controlled progression through four phases of the cell cycle: G1, S, G2, and M [210]. At the center of cell cycle regulation are cyclins (CDCs) and cyclin-dependent kinases (CDKs). CDKs control the progression from growth phases (G1 and G2) to phases associated with DNA synthesis (S) and mitosis (M) (Fig. 8.2). Expression of cyclins is cell cycle dependent and control is regulated through transcriptional, translational, and posttranslational mechanisms [211]; alternatively, the expression of CDKs remains fairly constant. Specific CDC-CDK complexes facilitate progression through the various cell cycle checkpoints. Briefly, in response to extracellular growth signals, cyclin D1 is synthesized and complexes with CDK4 which initiates progression through G1S. The cyclin D1-CDK4 complex phosphorylates RB, which dissociates from the E2F/DP1/RB complex allowing E2F-mediated transcription of cyclins A and E. Cyclin E complexes with CDK2 facilitate progression through G1S checkpoint. Cyclin A complexes with CDK2 to allow progression through S-phase and cyclin B then complexes with CDK1 to regulate G2M and entry into mitosis.
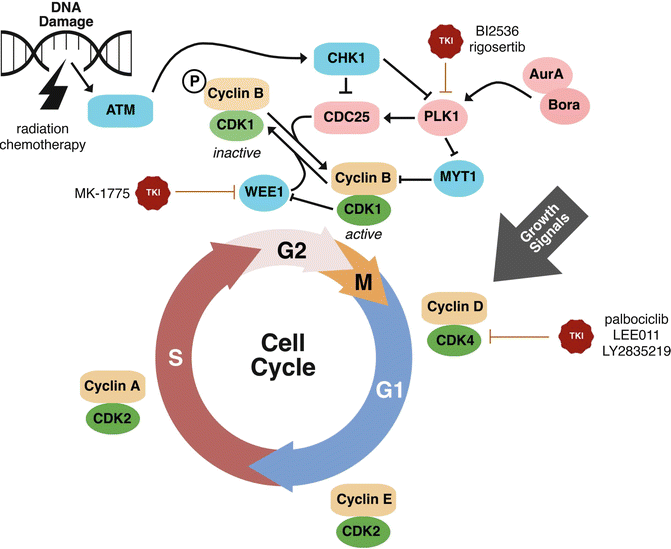
Fig. 8.2
Emerging molecular targets, including cell cycle regulatory targets, for HNSCC treatment. The cell cycle is a tightly regulated process in normal tissues. During tumorigenesis, cell cycle regulatory mechanisms are inhibited and stimulators of cell cycle are enhanced. Several emerging drugs targeting cell cycle regulatory proteins are in active investigation for the treatment of HNSCC. Promising targetable cell cycle regulatory proteins include PLK1, WEE1, and CDK4. Inhibition of CDK4 or PLK1 inhibits cell cycle. Inhibition of WEE1 leads to inhibition of the G2M checkpoint and, in the context of G1S checkpoint deficiency, promotes an accumulation of DNA damage and cellular catastrophe. These agents are of high promise given the potentially utility in a wide range of human malignancies
Unrestrained cell growth and proliferation are hallmarks of cancer [37]. Cellular functions such as apoptosis, senescence, differentiation, and proliferation are all integrally linked with the cell cycle regulatory machinery [212]. Alterations in this machinery facilitate the biological potential and aberrant behavior of cancer cells. Abrogating proliferative signaling by targeting specific driver proteins may provide new targets for therapeutic intervention in HNSCC. Traditional chemotherapies, such as antimitotics, form the mainstay chemotherapy in many solid tumor types. However, these agents are relatively indiscriminant in their effects on tumor and normal cells and adverse effects are significant. Agents that selectively target mitosis-regulating proteins that are differentially expressed in tumor (relative to normal tissues) could enhance the armamentarium of adjuvant treatments in HNSCC. Targets that fit this profile, in phase II of clinical development in HNSCC, include PLK1, WEE1, and CDK4/6 (Fig. 8.2).
8.3.1 PLK1
Polo-like kinases (PLKs1-5) are a family of highly conserved serine/threonine kinases related to the polo gene product of Drosophila melanogaster [213, 214]. PLKs play a complex and multifaceted role in orchestrating several steps of the cell cycle, which was recently reviewed in detail by Yim and Erikson [214]. PLKs are characterized by the presence of a conserved polo-box domain (PBD) in the C-terminal non-catalytic domain that controls targeting the kinase’s catalytic activity to specific binding partners and substrates [215]. The most well characterized PLK is PLK1. During the cell cycle, PLK1 activity is highest during late G2 and M phases and functions to regulate mitosis [216]. PLK1 drives mitosis through regulation and activation of CDK1, centrosome maturation, bipolar spindle formation, chromosome segregation, regulation of the anaphase-promoting complex, and execution of cytokinesis (Fig. 8.2) [216]. Because PLK1 permits passage through the spindle checkpoint, sustained or inappropriate PLK activation prevents checkpoint-mediated apoptosis [168]. PLK1 has also been shown to interact with other targetable cell cycle regulatory proteins such as WEE1 and AURKA [216].
PLK1 expression is normally the highest in tissues with the greatest turnover [216] and is highly expressed in several tumors types (e.g., breast, esophageal, lung, colon) and nearly all HNSCC [217–219]. The mRNA expression level of PLK1 is a negative prognostic indicator [217, 218]. Patients with high versus moderate tumor PLK1 expression had significantly shorter 5-year survival rates, and PLK1 mRNA expression levels correlate with advanced nodal status [218]. Although these data reflect only one HNSCC cohort, PLK1 expression patterns correlate with lower 5-year survival rates in lung and esophageal cancers [214].
PLK1 has been postulated to serve as a biomarker of proliferation (e.g., PCNA). However, ectopic expression of PLK1 was sufficient to transform NIH3T3 cells [220]. Transfection of antisense oligonucleotides targeting PLK1 mRNA in breast, cervical, and non-small cell lung cancer cells inhibited cell growth and reduced tumor growth in vivo [213]. In HNSCC cells and xenografts, PLK1 siRNA transfection reduced PLK1 mRNA expression and cell viability and induced apoptosis [219, 221]. These effects were enhanced by co-treatment with radiation [221]. In HNSCC cells BI2536, a selective PLK1 competitive inhibitor, reduced proliferation and induced G2/M arrest and apoptosis [222]. The dual pathway inhibitor rigosertib targets PI3K and PLK1, although there has been discussion as to whether rigosertib directly targets PLK1 [223]. Rigosertib elicited potent antiproliferative effects in 11/16 HNSCC lines evaluated, and sensitivity was confirmed in orthotopic xenografts [223]. Interestingly, all sensitive cell lines carried the combination of a PIK3CA-activating mutation and a p53-inactivating event [223]. Three out of eight tumors demonstrated sensitivity to rigosertib in patient-derived xenografts (PDX) [223], and two of the sensitive PDXs carried PIK3CA amplification. Although these results implicate biomarkers of sensitivity to the purported dual PI3K/PLK1 pathway inhibitor rigosertib, it is more likely that PLK1 pathway inhibition is a consequence of abrogated upstream signaling.
Rigosertib and BI2536 are the only PLK1 pathway inhibitors that have progressed to phase II studies [58]. In a phase I study in advanced solid tumors, rigosertib demonstrated clinical activity in two out of six HNSCC patients with one CR and one PR [224]. A phase II trial (NCT01807546) evaluating rigosertib as a monotherapy in R/M platinum-resistant squamous cell carcinomas, including HNSCC, is open and active [58]. The PLK1 inhibitor BI2536 demonstrated a single PR in one HNSCC patient in a phase I study, although the patient progressed after five additional courses [225]. However, a multicentered parallel phase II trial (NCT00526149) of BI2536 in patients with advanced R/M HNSCC (N = 14) and other cancers demonstrated no CR or PR; the best response was stable disease and dose-limiting toxicities were frequent [226]. The physiologic role of PLK1 in orchestrating cell cycle control and its prognostic significance in HNSCC support PLK1 as a rational target in the treatment of HNSCC; however, few clinical studies targeting PLK1 are currently active [58].
8.3.2 WEE1
p53 is the most commonly mutated gene in HNSCC with over 50 % of tumors harboring mutations [122]. p53 mutations are associated with decreased survival, metastasis, and resistance to radiation therapy. Given the biological significance of p53 in HNSCC, it is surprising that therapies targeting p53-inactive tumors have been elusive. One such potential target is WEE1. WEE1 is a tyrosine kinase that functions as a G2M checkpoint gatekeeper (Fig. 8.2). The activation of cell cycle checkpoints after DNA damage prevents cells from progressing to mitosis with damaged or un-replicated DNA [227]. Most cancer cells lack the G1S checkpoint due to deficient or mutated p53/pRb. These cells are also resistant to DNA damage sensing and p53-mediated apoptosis [228]. However, in p53/pRb-deficient cells, G2M checkpoint is typically maintained allowing DNA repair before mitotic entry [228]. The cyclin B-CDK1 complex tightly regulates G2M checkpoint and mitotic entry. At the beginning of S-phase, cyclin B expression is low and increases until it peaks at the end of G2. CDK1 expression remains uniform through the cell cycle. As cyclin B expression increases cyclin, B-CDK1 complexes form and are maintained in an inactive state through inhibitory phosphorylation of specific tyrosine residues by WEE1 and MYT1. Progression through G2M and entry into mitosis is initiated by dephosphorylation of the inactive cyclin B-CDK1 complex by the phosphatase CDC25. Activated cyclin B-CDK1 complex then inhibits its inhibitor (WEE1) and further activates CDC25 through a positive feedback loop. This initiation commits the cells to mitosis [228]. Checkpoint kinase 1 (CHK1) senses DNA damage, governs the DNA damage response, and controls the G2M checkpoint. In the presence of DNA damage, CHK1 inhibits CDC25, thus preventing CDK1 activity and mitotic entry [228]. Therefore, inhibition of WEE1 kinase activity permits CHK1-independent mitotic entry in the presence of DNA damage.
Enhanced or sustained DNA repair is one mechanism of resistance to DNA-damaging chemotherapeutics [229]. Abrogating G2M checkpoint in the context of G1S checkpoint deficiency promotes the rapid accumulation of DNA damage and cellular catastrophe [230]. Furthermore, this process is selective for tumor cells compared to normal cells, in part, due to maintained G1S checkpoint in normal cells. Identifying targetable proteins that regulate G2M, such as WEE1, may be one strategy to extend the utility of DNA-damaging chemotherapeutics. The experimental anticancer potential of WEE1 inhibition was first demonstrated through TKI and siRNA knockdown [228]. WEE1 knockdown elicited a 5-fold increase in adriamycin-induced apoptosis compared to controls in HeLa cells [228]. Similarly, the first WEE1-specific TKI, MK-1775, sensitized p53-deficient cells and xenografts to chemotherapeutics (i.e., gemcitabine, cisplatin, and 5-fluoruracil) in various human cancers [230, 231]. WEE1 expression is essential to error-free cell division, and thus, MK-1775 treatment leads to an accumulation of DNA damage and apoptosis independent of chemotherapeutics in vitro and in vivo [232]. In HNSCC, WEE1 was identified as a potential therapeutic target using a functional kinomic screen in primary and metastatic cell lines harboring p53 mutations [229]. Validation of targetable kinases with known TKIs in HNSCC demonstrated that MK-1775 exhibited the broadest activity in p53-mutant cells [229]. Treatment of HNSCC p53-mutant xenografts with MK-1775, or in combination with cisplatin, led to significant regression of tumors and a synergistic effect between the two treatments [229]. The levels of pWEE1 and pCDC2 in p53-mutant xenografts were reduced with MK-1775 treatment [229]. These results support the notion that WEE1 is a viable target in p53-mutated cancers and that enhanced antitumor activity can be achieved through coadministration of DNA-damaging agents [229]. Osman et al. (2015) published similar results in a study of HNSCC with high-risk p53 mutations; they demonstrated that nanomolar concentrations of MK-1775 sensitized cell lines and xenografts to cisplatin-induced senescence [233]. In the background of wild-type p53, treatment with MK-1775 and cisplatin did not provide significant improvement over cisplatin alone in vitro or in vivo [233].
There are currently 20 trials listed on the www.clinicaltrials.gov website evaluating MK-1775 [58]. Fifteen of these studies are active, one has been completed, and two have been terminated. Preliminary results from a completed phase I trial (NCT00648648) in advanced solid tumors reported promising findings [234]. Single-dose monotherapy with MK-1775 up to 1300 mg did not yield any dose-limiting toxicities (DLTs). However, patients receiving chemotherapy (gemcitabine, cisplatin, or carboplatin) and MK-1775 frequently experienced DLTs [234]. A single active phase II clinical trial (NCT02196168) is currently recruiting patients to evaluate cisplatin with or without MK-1775 in patients with R/M HNSCC [58]. More studies are necessary to thoroughly evaluate the role of WEE1 inhibitors in the treatment of HNSCC.
8.3.3 CDK4/6
Aberrantly regulated cell cycling and the subsequent deregulation of proliferation are trademarks of cancer [37]. The cyclin D/CDK4/RB network is often involved in this process through various mechanisms including: amplification or overexpression of CCND1, amplification or overexpression of CDK4, activating mutations in CDK4, RB loss, and inactivation of cyclin D-CDK4 complex inhibitors (e.g., p16) via methylation, deletion, or mutations [235]. In HNSCC, CCND1 is frequently amplified and the cyclin D-CDK4 inhibitor CDKN2A is frequently methylated or deleted. Collectively, deregulation of the cyclin D/CDK4/RB network is found in 93 % of HNSCC tumors [164, 236]. Furthermore, targeting deregulated proteins involved in the cell cycle is a logical anticancer strategy as these inhibitors would be broadly applicable to many cancers. CDK4/6 inhibitors have been in development for several years, and, thus far, candidate drugs had unacceptable toxicity, were nonselective, or lacked activity [209]. The recent emergence of selective CDK inhibitors with promising safety profiles has facilitated preclinical and clinical investigations of CDK inhibitors in various cancers, including HNSCC [209]. In summary, targeting members of the cyclin D-CDK4 complex, in the context of sustained RB function, may arrest cell cycle and thus is a promising target for the treatment of HNSCC.
The most clinically advanced CDK4/6 inhibitor is palbociclib [235]. Palbociclib, an experimental CDK4/6 inhibitor, has been granted priority review status by the FDA for first-line treatment of ER (+) HER2 (−) previously untreated breast cancer [209]. Palbociclib selectively inhibits the cyclin D1-CDK4, cyclin D2-CDK4, and cyclin D3-CDK6 complexes, thus preventing RB phosphorylation and degradation (Fig. 8.2) [237]. In a recently published dissertation examining 416 cancer cell lines derived from 12 distinct cancer sites (including HNSCC), sensitivity to palbociclib was predicted by ERBB2 and CCND1 amplification whereas resistance was conferred by CCNE1 amplification and RB deletion [238]. The most sensitive cell lines were from breast; interestingly HNSCC lines were comparably sensitive [238]. Effects in specific HNSCC cell lines were not reported and the study was not sufficiently powered to examine relationships between sensitivity to palbociclib and presence of specific genetic events in HNSCC [238]. Likewise, palbociclib inhibited the growth of several tumor types in vitro and in human tumor xenografts including liposarcoma, mantle cell lymphoma, myeloma, and breast, ovarian and colon cancers at nanomolar concentrations [210]. Not surprisingly, these effects were dependent on functional RB [210]. LEE011 is another selective CDK4/6 inhibitor that has demonstrated similar efficacy in cell lines and humor tumor xenografts of breast cancer [239], neuroblastoma [240], and liposarcoma [241]; specific effects in HNSCC have not been reported. P276-00, an experimental flavone with nonselective CDK inhibitor activity, inhibits the growth of several types of cancer cells including mantle cell lymphoma; breast, prostate, and lung cancers; melanoma; and HNSCC. In HNSCC cells and human tumor xenografts, P276-00 arrested cells in G1/S leading to reduced proliferation, p53-mediated apoptosis, and inhibition of CCND1, MYC, and E2F1 expression and other mitogenic proteins [242]. Although limited, the preclinical data for selective CDK4/CDK6 inhibitors suggest that CDK4/6 will remain a viable target in the treatment of HNSCC.
Early-phase clinical studies examining promising CDK inhibitors are currently underway. Three early-phase clinical studies have been completed with the nonselective CDK inhibitor P276-00 in advanced and R/M HNSCC alone or in combination with radiation therapy or chemoradiation; the results are not yet published [58]. Selective CDK4/6 inhibitors palbociclib, LEE011, and LY2835219 are in early phases of clinical development for a variety of cancer types. Only palbociclib is currently being investigated in a phase I/II (NCT02101034) with cetuximab in advanced and R/M HNSCC that is considered incurable [58]. Given the high-priority review for the treatment of specific types of breast cancer, it is likely that more preclinical and clinical studies will emerge to address the true utility of CDK4/CDK6 inhibitors in the treatment of HNSCC.
8.4 Targeting the Tumor Microenvironment
The majority of cancer research examines the genetic/epigenetic alterations and oncogenic signaling pathways that drive malignant transformation. Accordingly, most emerging molecular targeted therapies for HNSCC target the malignant epithelium. However, the last three decades have elucidated that malignantly transformed cells do not act independent of the host [243–246]. Alternatively, tumors behave as organs that co-opt the surrounding phenotypically and genetically normal tumor microenvironment (TME) to cooperate in their malignant schema (Fig. 8.3) [245]. The TME includes the surrounding mesenchyme (tumor-associated fibroblasts [TAFs] and extracellular matrix [ECM]), vasculature (blood and lymphatic vessels), and cancer-associated inflammatory infiltrate [243]. The intricate web of exploitation directed by tumor cells on the TME contributes considerably to tumor heterogeneity and response (or lack of response) to chemotherapies [246, 247]. Contributions of the TME to cancer progression and heterogeneity include: supporting proliferative signaling, escaping growth inhibition, defying programmed cell death, permitting mitotic immortality, de novo angiogenesis, stimulating invasion and metastasis, reprogramming energy metabolism, and evading immune destruction [244]. Interfering with the tumor-TME relationship may provide additional weapons to the armamentarium of targeted anticancer agents currently being investigated. Agents that target the TME currently under clinical investigation have been focused to perturb angiogenesis (e.g., VEGF and VEGFR), signaling from the tumor stroma (HGF) (discussed above) and tumor-associated inflammation. Herein we highlight the angiogenesis pathway and immunomodulatory targets that are ongoing interests for pathway-targeted strategies for the treatment of HNSCC (Fig. 8.3).
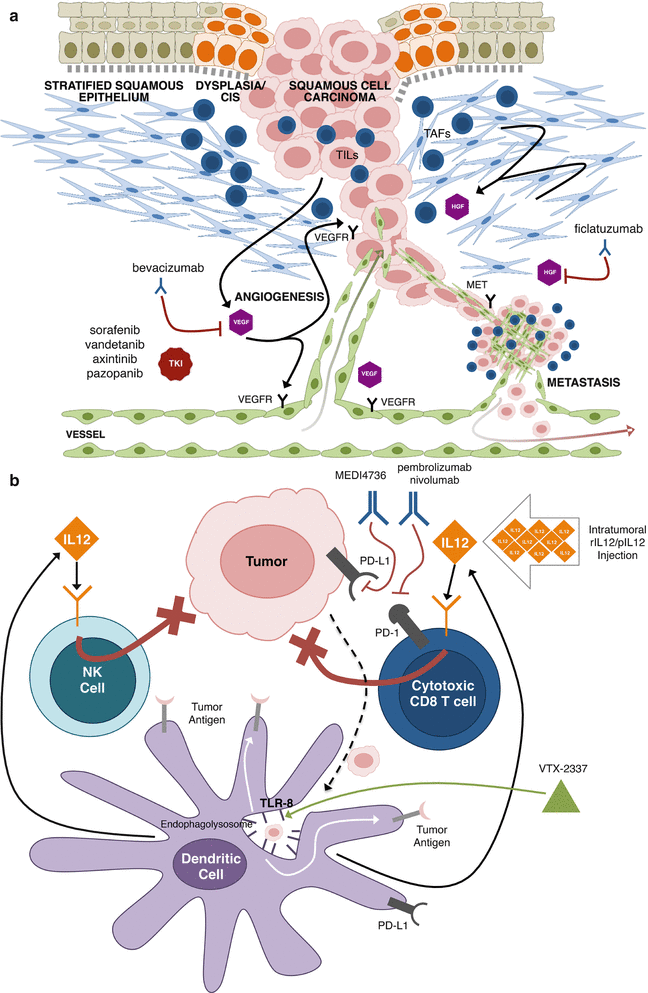
Fig. 8.3
Targeting the tumor microenvironment for the HNSCC treatment. (a) The tumor microenvironment includes the surrounding angiolymphatic vasculature, stroma, and infiltrating immune cells. Malignancies, including HNSCC, enlist the surrounding microenvironment (vasculature, tumor-associated fibroblasts [TAFs], tumor-infiltrating lymphocytes [TILs]) into a malignant schema. The figure above highlights the emerging molecular targets and their respective drugs that attempt to thwart this schema by targeting the tumor microenvironment (e.g., VEGF, VEGFR, HGF, MET). VEGF-sequestering mAbs “normalize” tumor vasculature and reduce vascular leakage and thus may enhance cytotoxic chemotherapies. Although it has been demonstrated that VEGFR is expressed on HNSCC cells, the vast majority of staining is in the tumor-associated vasculature. VEGFR-targeting agents also elicit profound effects on the vascular compartment, but may also inhibit the tumor compartment. Alternatively, HGF, a growth factor secreted by the tumor-associated fibroblasts, facilitates a more stem-cell-like phenotype in tumor cells and promotes invasion and metastasis. Like VEGF, sequestering HGF may prevent invasion and metastasis. The immune system plays an important, interdependent role in tumor establishment, growth, and immune evasion. (b) Tumors actively recruit and co-opt immune cells. Tumor cells, including HNSCC, express co-receptor PD-L1, whereas PD-1 is expressed on activated CD8 T cells. Co-receptor binding attenuates activated T-cell cytotoxicity and leads to T-cell exhaustion and apoptosis. Inhibitors of PD-1 or PD-L1 prevent this interaction and maintain activated cytotoxic CD8 T-cell tumor cell killing. Intratumoral IL-12 binds to IL-12 receptors on NK cells and cytotoxic CD8 T cells to enhance tumor cell killing. Tumor cells actively suppress dendritic cell presentation of tumor antigen and endophagolysosome activity. TLR-8 agonists (i.e., VTX-2337) enhance the phagocytic activity of dendritic cells, which engulf apoptotic tumor cells and present tumor antigen on the surface. Dendritic cell presentation of tumor antigen can then stimulate CD4 and CD8 T-cell activity and tumor immunity. Therefore, these agents act to reestablish the host’s immune response to tumor, thus potentially immunizing the host to tumor antigen
8.4.1 Angiogenesis
Over 40 years ago, it was demonstrated that tumor growth becomes severely restricted in the absence of ingrowth (<2 mm) of new blood vessels [248]. These seminal studies generated the hypothesis that targeting neovascularization might provide a therapeutic strategy for the treatment of solid tumor malignancies [248]. It is now well established that tumors require, and actively recruit, new vasculature to sustain growth, facilitate invasion, and permit metastatic spread [243, 249]. Angiogenesis is the de novo formation of blood vessels and is widely regarded as a hallmark of cancer [37, 245]. Strategies to target angiogenesis to treat human malignancies are in various phases of clinical use and clinical development [58]. It is postulated that inhibitors of angiogenesis target the endothelial compartment of tumors. Evidence also implicates that some angiogenesis inhibitors may also act directly on tumor cells. Thus, agents that target angiogenesis have tremendous promise for the treatment of HNSCC.
The vascular endothelial growth factor (VEGF) ligand family consists of six members including: VEGFA, VEGFB, VEGFC, VEGFD, and VEGFE and placenta growth factor (PGF) [250]. These ligands variably bind to three VEGF receptors (i.e., VEGFR1, VEGFR2, and VEGFR3) to regulate endothelial cell growth and differentiation [251]. Ligand binding activates the tyrosine kinase activity of these receptors to transduce specific angiogenesis-promoting effects [250]. VEGFR1 is expressed primarily in the vasculature and promotes endothelial cell migration but has limited effects on proliferation [251]. With a 10-fold stronger affinity for VEGFA, and reported antiangiogenic properties, it is proposed that VEGFR1 acts as a negative regulator of angiogenesis or as a decoy receptor to regulate VEGF concentrations [250, 251]. However, its exact role in HNSCC biology is not yet clear. Van Limbergen et al. reported that VEGFR1 was overexpressed in HNSCC cells and tumors and that overexpression reduced the sensitivity to radiation and promoted survival [252]. VEGFR2 is also expressed in the vasculature and is the predominant signal transducer for pathologic angiogenesis, mediating cell proliferation, survival, and motility [251, 253]. However, VEGFR2 has been reported to be expressed in HNSCC cells and VEGF-VEGFR2 signaling drives proliferation and survival [254
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


