Fig. 10.1
Model of the unfolded protein response
As increased basal levels of UPR signaling appear to characterize the malignant phenotype, we have hypothesized that small molecule enforcement of the UPR might be a productive anticancer approach [22–25]. The foundation of this hypothesis is rooted in the idea that a stress could be administered systemically to shift the balance of UPR signaling away from adaptation and toward apoptosis, specifically in malignant cells. Healthy cells (with low or no basal UPR activation), immediately adjacent to tumor cells, might respond to the systemic stress by mounting an effective (adaptive) UPR and return to homeostasis. In this chapter, we will discuss the rationale and feasibility of targeting the unfolded protein response (UPR) as a novel approach for a cancer therapy from the perspective of head and neck cancers our laboratory has been studying for the last 15 years.
10.2 NF-κB, Proteasome Inhibitors, and Endoplasmic Reticulum (ER) Stress
10.2.1 NF-κB
The landmark discovery that NF-κB signaling directly interfered with chemotherapy-induced apoptosis [26, 27] prophetically ushered in a new era of cancer drug discovery. Many pharmaceutical companies and academic chemical genomic centers simultaneously shifted their foci toward the development of gene therapy strategies and the identification of small molecules to inhibit NK-κB. The promise of potentiating tumor cell death by short-circuiting an ancient survival mechanism using less toxic drugs at (hopefully) lower doses ignited the promise of closing the outcome gap for HNSCC and myriad other malignancies [26, 28].
Under resting conditions, p65, a member of the NF-κB family of transcription factors, is held quiescent in the cytoplasm by the ankyrin repeat family protein IκBα, which binds to and masks the NF-κB nuclear localization signal. When TNFα binds to its cognate receptor or when a cell is exposed to a xenotoxic stress (e.g., chemotherapy), the dual phosphorylation of IκBα at serines 32 and 36 targets it for ubiquitination and degradation by the 26S proteasome (Fig. 10.2). Un-tethered NF-κB is then free to move into the nucleus to orchestrate a well-characterized survival program that includes early response genes and regulators of apoptosis and inflammation. For a comprehensive list with references, please see Boston University at http://www.bu.edu/nf-kb/gene-resources/target-genes/.
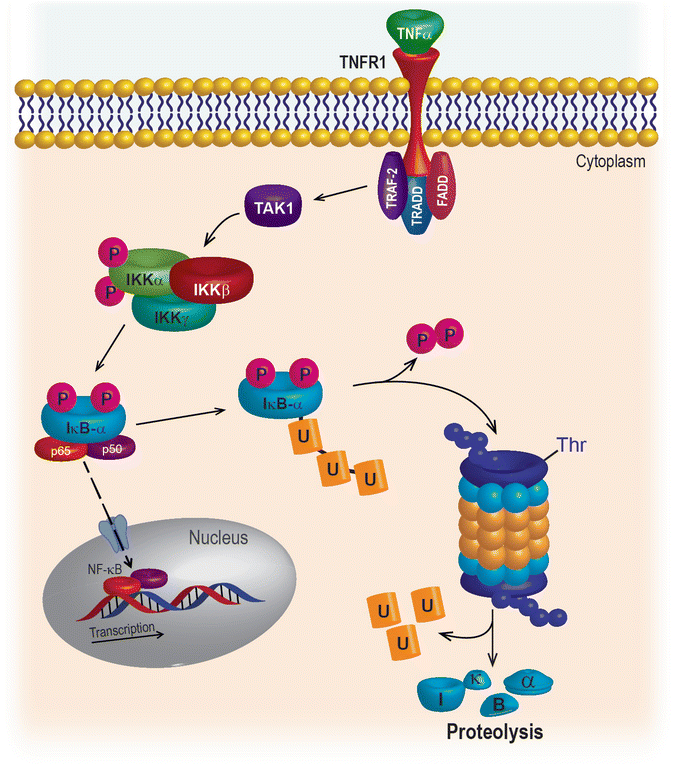
Fig. 10.2
NF-κB signaling pathway
Constitutively activated NF-κB signaling is a feature of squamous cell carcinomas of the oral cavity, head and neck, and larynx [29]. NF-κB regulates genes associated with early response, inflammation, immunity, stress response, and survival, all of which work together to drive aberrant proliferation, evasion of apoptosis and necrosis, and resistance to cancer treatment. Many strategies have been considered to inhibit the nuclear translocation of NF-κB family members to avert the aberrant survival of cancer cells. However, it has become clear that many oncogenic mutations occur upstream of NF-κB, and inhibition of nuclear translocation does not sufficiently sensitize malignant cells to chemotherapy or radiation. The upstream kinases (IKKs) that regulate NF-κB activation have also been targeted therapeutically; however, IKKs have been shown to govern inflammation and normal immune function that are critical to normal cell physiology. Although there is still tremendous effort in the search for effective direct inhibitors of NF-κB, to date, none have made it to the clinic for the treatment of HNSCC or any other cancer.
10.2.2 Proteasome Inhibitors
The 26S proteasome is responsible for the degradation of many signaling molecules, transcription factors, and tumor suppressors, including IκBα. During the mid- to late 1990s, it was hypothesized that proteasome inhibition might be a mechanism by which IκBα could escape degradation. It was thought that high cytosolic levels of IκBα might prevent NF-κB from translocating to the nucleus and enhance the cytotoxic effects of chemotherapy [28]. Bortezomib, also known as PS-341, was the first-in-class irreversible inhibitor of the chymotrypsin-like activity of the 26S proteasome [30–32] (Fig. 10.3). Early reports from studies in cell culture models indicated that bortezomib potentiated cell death in HNSCC and multiple myeloma, cancers known to be sensitive to chemotherapy following NF-κB inhibition [28, 33–35]. Millennium Pharmaceuticals made an unprecedented effort to collaboratively gather information and to discover other possible mechanisms of bortezomib-induced cell death. This pioneering effort involved sharing bortezomib at no cost to scores of academic laboratories all over the world. The results were staggering. As in vitro and in vivo data poured into the literature about dozens of cancers and thousands of genes that were affected by bortezomib, it became clear that Millennium’s collaborative effort would expedite the journey of bortezomib from bench to bedside, and it was approved in 2003 for patients with refractory multiple myeloma.
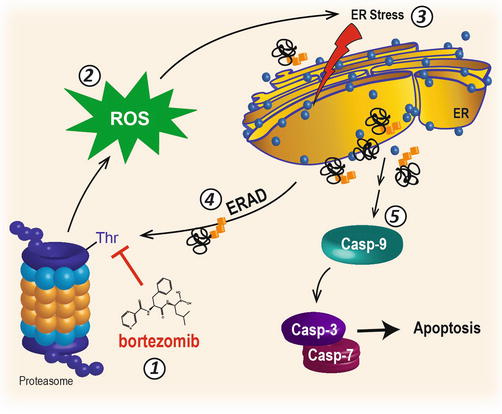
Fig. 10.3
Proteasome inhibition and ER stress
Early preclinical studies with PS-341 in an HNSCC cell culture model unexpectedly demonstrated that proteasome inhibition could induce apoptosis (IC50 ~ 500 nM) without the addition of NF-κB-inducing agents such as TNFα or other cytotoxic molecules [36]. Gene array analysis of HNSCC, multiple myeloma, and breast cancer cells treated with proteasome inhibitors revealed an accumulation of transcripts for genes associated with protein-folding machinery, activating transcription factor (ATF) family members, and the heat shock response [36–38], implicating ER stress as a potential mechanism of cell death. Earlier studies had in fact reported that lactacystin or small peptidyl aldehyde proteasome inhibitors (e.g., MG132) could increase protein transcripts for ATF proteins, HSP70 family members, glucose-regulated proteins (i.e., GRP78/BiP and GRP94), and CHOP, all of which are integral to the UPR. Although proteasome inhibitors have been shown to potentiate the cytotoxic effect of chemotherapy and radiation in a variety of cell lines, including HNSCC [35, 39–42], the realization that PS-341 could induce ER stress and UPR-dependent cell death [35, 36, 43–45] underscored a new hypothesis in that ER stress and the UPR might be targeted therapeutically for HNSCC and other cancers.
10.2.3 ER Stress and the UPR
It has been estimated that approximately one-third of all proteins manufactured by a typical cell are posttranslationally modified (i.e., with N- and O-linked glycosylation, disulfide bond formation, sulfation, phosphorylation, and proteolytic cleavage) and folded into an operative three-dimensional form in the endoplasmic reticulum (ER) [46]. Proteins that are secreted (e.g., growth factors and chemokines) and destined for lysosomes or that reside in the cell membrane (e.g., transporters, anchors, receptors and other enzymes) all undergo modification and folding in the ER. With the onerous burden of maintaining cellular function and homeostasis, it is essential that the ER not only possess efficient folding machinery and quality control but it must also have a system to address fluctuating folding demands. Specifically, the ER must be able to deal with situations where the demand for new proteins outpaces its current folding capacity.
The UPR is a homeostatic response to the accumulation of unfolded or misfolded proteins in the lumen of the ER. Perturbations in calcium homeostasis, redox status, or that affect glycosylation can interfere with protein folding. Inositol-requiring enzyme 1 alpha (IRE1α), activating transcription factor 6 (ATF6), and PKR-like ER kinase (PERK) are three ER transmembrane sensors that continually monitor folding and communicate disruptions to the cytoplasm to initiate the UPR (Fig. 10.1) [47–50]. Molecular chaperones (e.g., heat shock proteins (HSP) and glucose-regulated proteins (GRP) are of the first moieties that intercede to facilitate protein folding by shielding exposed unfolded regions of nascent peptides [51]. Under homeostatic conditions (unstressed), glucose-regulated protein 78/binding immunoglobulin protein (GRP78/BiP) is bound to the ER lumenal domains of PERK and ATF6 and thought to maintain them in an inactive state. When misfolded proteins begin to accumulate, GRP78/BiP is titrated away from these two sensors to increase folding productivity and initiate the UPR [47, 51, 52]. Subsequently, if lumenal proteins fail to fold, they are monoglycosylated by (UDP)-glucose:glycoprotein glucosyltransferase (UGGT), which targets them as substrates for the calnexin/calreticulin refolding cycle [53]. If a protein fails the lectin quality control cycle, it is trimmed by α1,2-mannosidases and directed toward the 26S proteasome through ER-associated degradation (ERAD).
PERK activation involves auto-oligomerization and auto-phosphorylation [54, 55]. Active PERK phosphorylates serine 51 of the α-subunit of the eukaryotic translation initiation factor 2-alpha (eIF2α) resulting in a brief global translation attenuation [47, 52]. eIF2α phosphorylation immediately allows the cell to remarshal its folding resources toward an adaptive response and avert death. ATF4 and CHOP increase following eIF2α phosphorylation and can induce the expression of a number of stress-dependent proteins, including death receptor 5 (DR5), tribbles-related protein 3 (TRB3), growth arrest and DNA damage-inducible protein 34 (GADD34), and NOXA [43, 56]. GADD34 is responsible for restoring global protein translation by dephosphorylating eIF2α to reinitiate translation. When translation is not resumed in a relatively quick and timely fashion, and the nuclear pools of ATF4, CHOP, and NOXA remain high, the balance of the UPR is shifted toward apoptosis [43, 57].
ATF6 is the least well characterized (mechanistically) of the three folding sentries, due largely to the chronically poor antibodies that are commercially available. ATF6 is a type II transmembrane protein transcription factor that spans from the ER lumen into the cytosol. The accumulation of misfolded proteins and the dissociation of GRP78/BiP from the lumenal domain prime ATF6 for loading into COPII vesicles and transport to the Golgi [58, 59]. Once at the Golgi, ATF6 is proteolytically cleaved N-terminally (membrane-lumen junction) by site one (S1) protease and C-terminally (membrane-cytosol junction) by site two (S2) protease. Free ATF6 then translocates to the nucleus where it binds to the ER stress element (ERSE) of UPR target genes. Molecular chaperones (e.g., GRP78/BiP, GRP94, and calreticulin) and folding proteins like protein disulfide isomerase (PDI) that enhance protein folding and increase the production capacity of the ER are all ATF6 targets [52, 60–65].
A hallmark of ER stress is the splicing of X-box-binding protein 1 (XBP1) mRNA following the activation of IRE1α. IRE1α was originally believed to be activated by a mechanism similar to ATF6 and PERK (i.e., by dissociation of GRP78/BiP from a lumenal domain) but was later found to be activated through the interaction of its conserved lumenal core domain with unfolded proteins [66, 67]. In mammals there are two IRE1 isoforms: IRE1α is expressed globally and IRE1β is found exclusively in the respiratory and gastrointestinal tracts [52]. Upon binding to unfolded proteins, IRE1 autophosphorylates and oligomerizes in the ER membrane. Activated IRE1α has intrinsic exoribonuclease activity in the cytosol that facilitates the splicing of a 26 base intron from XBP1 (or XBP1u to denote un-spliced). The biological role of the XBP1u, if there is one, has remained elusive. Religated XBP1encodes a basic leucine zipper (b-ZIP) transcription factor that regulates adaptive and apoptotic UPR target genes. XBP1 targets include HSPs and other ER-localized chaperones (e.g., GRP78/BiP) [68], foldases [47, 49–52], ER-associated degradation protein (EDEM), ER-localized Dnajb9 (Dnaj4), and protein disulfide isomerase (PDI) [47, 52, 68]. Interestingly, XBP1−/− murine embryonic fibroblasts (MEFs) treated with tunicamycin demonstrated reduced expression of proapoptotic molecules CHOP and GADD45 [68] indicating that the IRE1-XBP1 axis of the UPR might also contribute to cell death.
The outcome of UPR signaling can seem very enigmatic considering the different roles of PERK, ATF6, and IRE1α. Cells can be protected by enhancing specific elements of the UPR that can alleviate ER stress and reestablish homeostasis while at the same time experience dramatically increased levels of cell death proteins. Several studies have demonstrated that the point at which a cell transition from adaptation to destruction is dependent upon the length of time each of the individual UPR branches are activated [57, 69, 70]. Taken together, these studies suggest that prolonged IRE1 signaling and enhanced production of chaperones improves cell survival, but with continuous PERK activation, chronically high levels of ATF4 and CHOP, and persistent eIF2-α phosphorylation, a cell is doomed to an apoptotic fate. Another key fate-determining factor is the length of time translation is paused. In a classic study, Scheuner et al. demonstrated that when eIF2α cannot be phosphorylated, translation is uninterrupted during ER stress, and cells die very early when challenged with UPR-inducing stimuli [71]. Another more recent paper from the Kaufman group demonstrated that when eIF2α was not rapidly dephosphorylated by factors downstream of ATF4-CHOP signaling (e.g., growth arrest and DNA damage-inducible protein 34 [GADD34]), a prolonged translational pause led to cell death [57]. In fact, the decisions that determine the balance between life and death during the UPR are not entirely known, and this remains an active and exciting field of investigation.
10.2.3.1 GRP78/BiP/HSPA5 in HNSCC
Glucose-regulated protein 78 kDA (GRP78), also known as binding immunoglobulin heavy-chain protein (BiP) or HSPA5, is an ATP-dependent HSP70 family member constitutively expressed in the ER lumen of most cells [72, 73]. GRP78/BiP is a multifaceted protein that plays a fundamental role in the maintenance of cellular and tissue homeostasis under physiologic and pathologic conditions. GRP78 was initially isolated from cultured cells grown in medium deprived of glucose [74]. BiP was independently discovered a few years later bound to the immunoglobulin heavy chains of pre-B cells [75], and a later study with normal rat liver samples determined GRP78 and BiP were identical [76]. The principal function of GRP78/BiP is to monitor and chaperone the folding of newly synthesized proteins in the ER. GRP78/BiP also participates in protein translocation into and out of the ER, the identification and degradation of proteins that fail to achieve a functional geometry, calcium storage, and the sensing and response to aberrant folding conditions [77]. GRP78/BiP was the first eukaryotic ER chaperone to be identified and, like all known HSP70 proteins, is ATP-dependent. ATP-bound GRP78/BiP has an “open” configuration that allows it to associate with proteins that have failed to fold. When hydrolyzed, the ADP bound form binds tightly to the misfolded proteins, which cannot be released until ATP is reintroduced to the nucleotide-binding cleft [78, 79]. Another interesting structural feature of GRP78/BiP and other ER-associated calcium-binding proteins (e.g., GRP94, calnexin and calreticulin) is the Lys-Asp-Glu-Leu (KDEL) ER-localization signal sequence. KDEL has been very useful as an ER marker for immunohistochemical (IHC) studies and subcellular fractionation studies. In concert with other calcium-binding KDEL proteins, GRP78/BiP modulates lumenal calcium homeostasis [80].
In addition to its fundamental role as a folding chaperone, GRP78/BiP facilitates the transport of new proteins across the ER membrane and regulates proliferation, tumor progression, angiogenesis, autophagy, chemoresistance, and apoptosis [81–83]. For a concise review, please see Fu and Lee 2006 [84]. A role for GRP78/BiP in cancer was first noted in B/C1ME fibrosarcoma cells stably transfected with amplified copies of antisense grp78. Inhibition of grp78 expression did not affect the growth of cells in culture but dramatically sensitized them to ER stress-mediated cell death. This landmark study also reported that the fibrosarcoma cells with grp78 deficiency either failed to form xenograft tumors in a murine model or created very small tumors that were quickly resorbed [85]. Increased GRP78/BiP expression in a wide variety of tumors suggests that GRP78/BiP may play a significant role in both normal and malignant tissue. Oftentimes, there are increased levels of misfolded proteins and ER stress in cancer cells that are the result of gene mutations and the stressful milieu of the tumor microenvironment (TME). GRP78/BiP has been characterized as an excellent and much needed biomarker of cancer progression. Elevated GRP78/BiP in patient samples has been associated with more aggressive cancers and poor survival in myriad cancers, including esophageal adenocarcinoma [86–89] and HNSCC [90–95]. The full utility of using GRP78/BiP expression as a predictive marker of HNSCC at the time of diagnosis to aid in both prognosis and determination of an appropriate treatment strategy is just coming to light. In order to do this, it is important to recognize the different roles that GRP78/BiP plays in tumor progression in different cancer cell types.
Increased expression of GRP78/BiP is a well-characterized biomarker of stress in the ER [96] and is considered a master regulator of ER homeostasis. Many studies have demonstrated a central role for GRP78/BiP in tumor progression and chemoresistance. The proprietary free software program Oncomine that interfaces with archived publically available gene array data was queried to examine GRP78/BiP expression in oral squamous cell carcinomas and normal tissue. Oncomine contained eleven gene array studies that included at least five paired HNSCC subtypes and five nonmalignant controls. GRP78/BiP was increased in all seven studies between 1.2- and 2.7-fold. A study-level meta-analysis confirmed that HNSCC cells displayed significantly elevated GRP78/BiP gene expression compared to normal tissue (P < 0.000001, single sample t-test) (Table 10.1). Immunohistochemical analysis of GRP78/BiP was performed with archived formalin-fixed paraffin embedded (FFPE) surgical samples from patients with neck, laryngeal, and lip squamous cell carcinomas with approval from the Karmanos Cancer Institute Protocol Review and Monitoring Committee and the Wayne State University Institutional Review Board. GRP78/BiP expression was consistently very low or absent in adjacent nonmalignant regions of the tissue sections, was increased in well-defined transition zones, and most intense in regions of malignancy (Fig. 10.4a, b). Control experiments performed with a BiP-specific blocking peptide demonstrated that the observed immunostaining was specific (Fig. 10.4c).
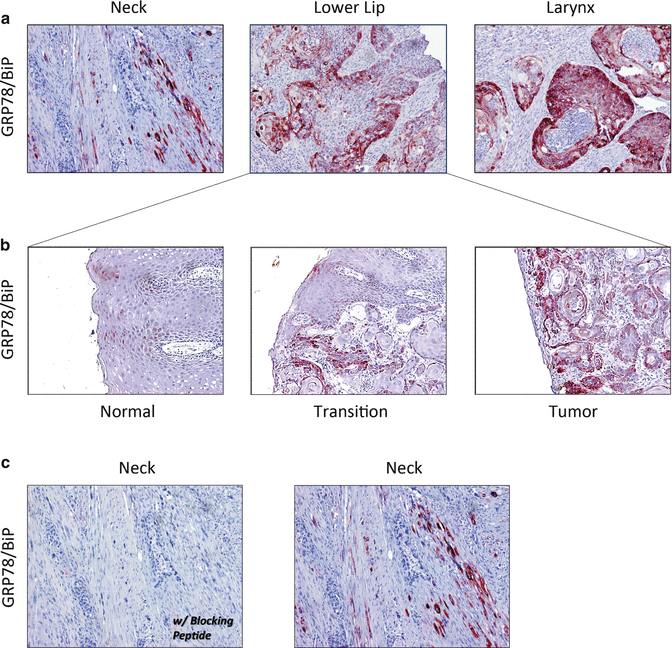
Table 10.1
GRP78/BiP expression in oral squamous cell carcinomas and normal tissue meta-analysis
|
Study
|
Cromer
|
Peng
|
Estilo
|
Ye
|
Talbot
|
Ginos
|
Sengupta
|
Pyeon
|
Kuriakose
|
Pyeon (oropharynx)
|
Pyeon (oral cavity)
|
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Fold
|
1.496
|
1.491
|
2.66
|
2.031
|
2.329
|
1.719
|
1.266
|
2.242
|
1.23
|
1.496
|
1.591
|
|
P value
|
0.01
|
7.69E–06
|
2.08E–09
|
1.45E–05
|
1.08E–08
|
1.37E–08
|
2.10E–02
|
6.26E–04
|
2.70E–02
|
1.84E–05
|
3.62E–04
|
|
Cancer samples (n)
|
34
|
57
|
31
|
26
|
26
|
41
|
31
|
15
|
22
|
6
|
4
|
|
Normal samples (n)
|
4
|
22
|
26
|
12
|
31
|
13
|
10
|
22
|
22
|
5
|
9
|
Fig. 10.4
Immunohistochemistry demonstrating GRP78/BiP expression in OSCC (a); expanded view of lower lip section to demonstrate increased GRP78/BiP staining with malignancy (b); and GRP78/BiP blocking peptide (left) to demonstrate antibody specificity
BiP promotes tumor cell survival and metastasis through the modulation of apoptosis and angiogenesis. Two recent studies suggested that GRP78/BiP might also represent a molecular target to kill refractory HNSCC stem cells [97, 98]. It has been well documented that patients with elevated levels of GRP78/BiP in their tumors display a higher pathologic grade, poorer prognosis, and an earlier relapse for many cancers including HNSCC. In an immunoblot analysis study of 204 patients, Lin et al. reported that GRP78/BiP overexpression was correlated with more aggressive potential in oral lesions. They found hyperexpression in 14 % of leukoplakias, 27 % of erythroplakias, 50 % of verrucous lesions, and 74 % of oral cancers (p < 0.0001) [91]. Conversely, another study reported that a decrease in GRP78/BiP protein expression was correlated with advanced tumor stage and neck lymph node metastasis [99]. However, it should be noted that the latter study was performed using only two oral squamous carcinoma cell lines from patients with high- or low-grade disease. Determination of GRP78/BiP protein levels with IHC studies using patient biopsies might enable clinicians to better stratify risk for patients with premalignant oral lesions.
A variety of heat shock proteins and translation initiation factors associated with the UPR have been demonstrated to be increased, and even predictive of recurrence, in head and neck and thyroid cancers [15, 16, 20, 86]. Increased GRP78/BiP (and markers of the UPR in general) in malignant cells likely reflects an attempt by the cancer to accommodate an increased demand to produce protein factors that regulate aberrant growth and compensate for a hypoxia and nutrient-deprived environment and to produce the many secretory chemokines and growth factors that characterize the malignant phenotype. GRP78/BiP expression is a feature of HNSCC, and there is a growing body of evidence that its expression correlates with tumor stage and is an indicator of chemoresistance [96, 100, 101]. These studies embolden the hypothesis that increased UPR in malignant tissue might be exploited pharmacologically to reduce tumor burden.
10.2.3.2 UPR Activation Versus Inhibition as an Approach to Cancer Therapy
The tumor milieu is host to a variety of inhospitable microenvironments that must be offset for malignant cells to proliferate and invade the host. Tumor expansion is hampered with poorly vascularized cells experiencing hypoxia, nutrient deprivation, and fluctuations in pH. A recent study reported that hypoxia-induced extracellular acidosis diminished p-glycoprotein activity thereby reducing the effectiveness of daunorubicin [102]. This is an important example of how the products of the metabolism and inner workings of a tumor cell can influence the extracellular environment and facilitate tumor survival in the face of treatment. As the growth of a tumor exceeds the capacity of the vasculature to supply sufficient oxygen and nutrients, a variety of stress responses are engaged, including the UPR. Since the UPR can be lethal or protective, it is reasonable to consider whether its activation or inhibition might be a more effective anticancer approach.
There are numerous preclinical studies that suggest that UPR inhibition might provide a suitable therapeutic option for some cancers, including HNSCC. Almost 20 years ago in a landmark paper, Jamora et al. observed that GRP78/BiP expression was induced in progressively growing neoplastic cells. To study the role of GRP78/BiP in tumorigenesis, they engineered fibrosarcoma cells that stably expressed a grp78 antisense construct. They found that the proliferation rates of the resultant cell lines were unchanged (in vitro); however, in vivo studies demonstrated that the cells were either incapable of making xenografts or made very small tumors that were eventually resorbed [85]. A later study by Langer et al. found that GRP78/BiP expression was increased in adenocarcinomas of the esophagus [86], and Lin et al. concluded that GRP78/BiP is a prognostic factor in oral precancerous lesions and was positively associated with oral cancer progression [91]. Taking advantage of the fact the tumor cells tend to be glucose starved with increased GRP78/BiP, it is plausible that interfering with GRP78/BiP might prevent the production of proteins (e.g., VEGF) required for vascularization. Chronic UPR inhibition and diminished blood vessel formation could serve to further asphyxiate tumor cells, deprive them of nutrients, and force tumor cells out of their aberrant cell cycle and induce a quiescent (i.e., dormant) state or perhaps induce apoptosis.
Small molecules that interfere with IRE1α and XBP1 have been reported to interfere with leukemia cell proliferation and multiple myeloma, respectively. Blocking the IRE1α-XBP1 axis of the UPR might be predicted to decrease levels of transcriptional targets of XBP1, such as GRP78/BiP and other heat shock proteins and foldases needed to restore homeostatic protein folding. However, there are several reports that inhibition of IRE1α-XBP1 signaling with small molecules increases ER stress as evidenced by eIF2α phosphorylation and the accumulation of GRP78/BiP and CHOP [103–105]. Compounds that inhibit XBP1 and activate CHOP are particularly attractive from an anticancer perspective in that CHOP transcripts downregulate BCL2, increase reactive oxygen species, and induce expression of genes that reinitiate protein synthesis following ER stress, all of which enhance the apoptotic arm of the UPR [57, 106]. These observations have been limited to preclinical studies in human cancer and rat kidney studies and have not yet been evaluated in clinical trials [104, 107].
Many FDA-approved cancer drugs, none of which were developed to target the UPR, have been reported to enhance ER stress and rely on the UPR to exert a cytotoxic effect (Table 10.2).
Table 10.2
Preclinical compounds and FDA-approved drugs demonstrating UPR activation
|
Drug/compound
|
Classification
|
Clinical/preclinical status
|
FDA approval (year)
|
Drug indications
|
Diseases studied
|
UPR indications
|
Reference
|
|---|---|---|---|---|---|---|---|
|
B-19
|
(Curcumin analog)
|
Preclinical
|
Novel chemotherapeutic agent
|
Ovarian cancer
|
Upregulated protein levels of BiP, XBP-1, ATF6, and CHOP, induced cleavage of caspase-3
|
Qu et at. [108]
|
|
|
Bevacizumab (Avastin)
|
rhuMAb (biologic)
|
Phase III/IV
|
2014
|
Inhibits VEGF receptor binding
|
Breast cancer, colorectal cancer, neuroendocrine tumors, non-small cell lung cancer, ovarian cancer, pancreatic cancer, prostate cancer, renal cell carcinoma, small cell lung cancer
|
Upregulated ADAM17 (PERK/ATF4 and ATF6 regulated gene), TNFα, and TNFR
|
|
|
Borrelidin (Streptomyces sp.)
|
Natural product
|
Preclinical
|
Angiogenesis inhibitor, threonyl-tRNA synthetase inhibitor
|
ALL, breast cancer fibrosarcoma, HNSCC osteosarcoma
|
Activated GCN2 pathway, phosphorylation of eIF2α, upregulated CHOP protein levels
|
||
|
Bortezomib (Velcade) (PS-314)
|
Small molecule
|
Phase I/II/III/IV
|
26S proteasome inhibitor
|
APL, HNSCC, lymphoma, multiple myeloma, nasopharyngeal carcinoma, neuroblastoma
|
NF-κB inhibitor, induced caspase activation, upregulated BiP, phospho-PERK, ATF4, CHOP, and GADD34
|
||
|
Cantharidin (Meloidae (Spanish fly))
|
Natural product
|
Phase I
|
Topical wart medication
|
HNSCC
|
Induced splicing of XBP1 mRNA, upregulated phosphorylation of eIF2α, increased protein expression of ATF4 and CHOP, induced caspase activation
|
Xi et al. [23]
|
|
|
Carfilzomib (Kyprolis)
|
Small molecule
|
Second-in-class Phase I/II/III
|
2012
|
Irreversible 20S proteasome inhibitor
|
ALL, hematologic malignancies, HNSCC, lymphoma, mantle cell lymphoma, multiple myeloma, neuroendocrine cancer prostate cancer, small cell lung cancer, solid tumors
|
Induced phosphorylation of PERK and eIF2α, upregulation of ATF4 and BIK, activation of caspase-3
|
|
|
Celastrol (Tripteryguim wilfordii (Thunder of God vine) and Celastrusregelii)
|
Natural product
|
Preclinical
|
Anti-inflammatory and antioxidant properties, RA, SLE, psoriasis, nephropathy, and ankylosing spondylitis
|
OSCC
|
Induced splicing of XBP1 mRNA and upregulation of UPR genes (CHOP, GADD34, ATF3, ERDJ4, HERPUD1 and p21); increased protein levels of GRP78/BiP, ATF4, and CHOP; increased ubiquitinated proteins; increased NOXA; induced caspase activation
|
||
|
Celecoxib (Celebrex)
|
Small molecule
|
Phase I/II/III/IV
|
2001
|
Nonsteroidal anti-inflammatory drug, COX-2 inhibitor, VEGF activator
|
Breast cancer, colorectal cancer, esophageal cancer, HNSCC, pancreatic cancer, urothelial carcinoma
|
Upregulated GRP78, XBP1, and CHOP
|
|
|
Curcumin (Diarylheptanoid)
|
Natural product
|
Phase I/II/III
|
Anti-inflammatory properties
|
Breast cancer, CLL, colorectal cancer, endometrial cancer, HNSCC, non-small cell lung cancer, osteosarcoma, pancreatic cancer, prostate cancer, solid tumors
|
Phosphorylation of PERK and IRE1α, increased protein expression of CHOP, GADD34, p50-ATF6, and XBP-1
|
||
|
Dasatinib (Sprycel)
|
Small molecule
|
Phase I/II/III
|
2010
|
Inhibitor of SRC family protein-tyrosine kinases
|
ALL, CML, HNSCC, lymphoblastic lymphoma
|
Induced AMPK-dependent ER stress, depletion of intracellular Ca2+, induced expression of GRP78 and CHOP
|
|
|
DIM (3,3′-diindolylmethane)
|
Phytochemical
|
Phase I/II/III
|
Recurrent respiratory papillomatosis
|
Breast cancer, carcinoma, cervical cancer, oral squamous cel,l ovarian cancer, pancreatic cancer, prostate cancer
|
Akt-NF-κB, MAPK, and p53 signaling; upregulation of DR5; induced expression of BiP, IRE1α, and CHOP; and phosphorylation of p53 at Ser-15
|
||
|
Disulfiram (Antabuse)
|
Small molecule
|
Phase I/II/III
|
1951
|
Chronic alcoholism
|
Glioblastoma, head and neck cancer, melanoma, non-small cell lung cancer, prostate cancer, solid tumors (liver)
|
Induced splicing of XBP1 mRNA, induced ROS, upregulated phosphorylation of eIF2α, 26S proteasome inhibitor
|
|
|
Emodin
|
Natural product
|
Preclinical
|
Type 2 diabetes
|
Tongue cancer, SCC, colon cancer
|
Oxidative stress induced p53, upregulated BiP and CHOP
|
||
|
Entinostat (MS-275)
|
Synthetic benzamide derivative
|
Phase I/II/III
|
HDAC inhibitor
|
Breast cancer, kidney cancer, nasopharyngeal carcinoma, non-small cell lung cancer
|
Increased expression of GRP78
|
Baumeister wt al. [138]
|
|
|
Erlotinib (Tarceva)
|
Quinazoline derivative
|
Phase I/II/III/IV
|
2013
|
Inhibitor of EGFR tyrosine kinase activity
|
Colorectal cancer, esophageal squamous carcinoma, HNSCC, non-small cell lung cancer, squamous cell lung cancer
|
Increased spliced XBP1, p-eIF2α, ATF4, and CHOP protein accumulation
|
|
|
Flavokawain B (FKB)
|
Natural product
|
Preclinical
|
Novel chemotherapeutic agent
|
OSCC
|
Increased expression of HSP70
|
||
|
GL63
|
Curcumin analog
|
Preclinical
|
Novel chemotherapeutic agent
|
Hepatocellular carcinoma, lung cancer, nasopharyngeal carcinoma
|
Upregulated XBP1, ATF4, CHOP, Cox-2 inhibitor
|
||
|
HMJ-38
|
Small molecule
|
Preclinical
|
Angiogenic suppressor
|
Leukemia, oral carcinoma
|
Induced DNA fragmentation; increased intracellular Ca2+; increased BiP, p-eIF2α, CHOP, and ATF6 protein accumulation; induced cleavage of PARP and caspases
|
Lu et al. [146]
|
|
|
Ibrutinib (Imbruvica) (PCI-32765)
|
Small molecule
|
Phase I/II/III
|
2014
|
Bruton’s tyrosine kinase inhibitor
|
CLL, lymphoma, mantle cell lymphoma, multiple myeloma, non-small cell lung cancer
|
AKT and NF-κB inactivation, enhanced DNA damage, phosphorylation of eIF2α
|
Dasmanhapatra et al. [147]
|
|
Imatinib mesylate (Gleevec)
|
Small molecule
|
First-line treatment (phase I/II/III/IV)
|
2012
|
CD117 positive GIST tyrosine kinase inhibitor
|
Adenoid cystic carcinoma, ALL, AML, CML, GIST, mesothelioma
|
Increased BiP and CHOP protein accumulation
|
Du et al. [148]
|
|
Ixazomib (MLN9708)
|
Small molecule
|
Second generation (phase I/II/III)
|
Proteasome inhibitor
|
ALL, AML, follicular lymphoma, multiple myeloma
|
Activation of BiP, eIF2α, and CHOP, inhibition of NF-κB
|
Chauhan et al. [149]
|
|
|
Lirodenine
|
Natural product
|
Preclinical
|
Novel chemotherapeutic agent
|
Laryngocarcinoma, ovarian adenocarcinoma
|
Upregulated p53, induced DNA fragmentation, induced caspase-3 cleavage
|
||
|
ML-291 (sulfonamidebenzamide)
|
Small molecule
|
First-in-class chemical probe
|
Selectively activates the apoptotic, but not the adaptive, arm of the UPR
|
HNSCC
|
Induced splicing of XBP1 mRNA, upregulated CHOP and GADD34 gene expression
|
||
|
Nelfinavir mesylate (Viracept)
|
Small molecule
|
First-line treatment (phase I/II/III)
|
1997
|
HIV antiretroviral proteasome inhibitor
|
Cervical cancer, gastric cancer, lymphoma, multiple myeloma, non-small cell lung cancer, pancreatic cancer SCC
|
Increased protein levels of phospho-eIF2α and ATF3, induced activation of caspase and PARP
|
Gills et al. [153]
|
|
NH2-PS
|
Polystyrene nanospheres
|
Preclinical
|
Imaging and drug delivery applications
|
Lung epithelial
|
Increased ROS generation, induced misfolded protein aggregation and ER stress, upregulated IRE1α protein levels
|
Chiu et al. [154]
|
|
|
NMS-873
|
Small molecule
|
Preclinical
|
Allosteric inhibitor of the VCP/p97
|
Colorectal carcinoma
|
Increased protein expression of BiP, CHOP, and GADD34, induced activation of caspase-3 and PARP
|
Magnaghi et al. [155]
|
|
|
Oprozomib (ONX 0912)
|
Bioavailable derivative
|
Phase I/II
|
Irreversible 20S proteasome inhibitor
|
Hepatocellular carcinoma, HNSCC, multiple myeloma
|
Induced phosphorylation of PERK and eIF2α, upregulation of ATF4 and BIK, activation of caspase-3
|
||
|
Patulin (Penicillium sp., Aspergillus sp. and Byssochlamys sp.)
|
Natural product
|
Preclinical
|
Potassium uptake inhibitor
|
Nasopharyngeal carcinoma, OSCC
|
Upregulation of BiP, ATF4, CHOP, ATF3, and GADD34 gene expression, induced XBP1 mRNA splicing, upreglated CHOP protein expression
|
||
|
Pemetrexed (Alimta)
|
Small molecule
|
First-line treatment
|
2012
|
Inhibits thymidylate synthase
|
Adenocarcinoma, ALL, head and neck cancer, malignant pleural mesotheliom,a ovarian cancer, thyroid cancer
|
Upregulation of ATF4, ATF3, and Noxa protein levels, induced activation of caspases and PARP
|
|
|
Polydatin
|
Natural product
|
Preclinical
|
Natural precursor of resveratrol
|
Nasopharyngeal carcinoma
|
Upregulation of XBP1s, ATF, and CHOP protein expression, induced caspase activation
|
Liu et al. [159]
|
|
|
Quinone methide aurin
|
Small molecule
|
Preclinical
|
Heat shock response inducer
|
Malignant melanoma
|
Increased levels of CHOP mRNA, upregulated protein expression of CHOP, increased phosphorylation of PERK and eIF2α
|
Davis et al. [160]
|
|
|
Resveratrol
|
Natural product
|
Phase I/II
|
Antioxidant, anti-inflammatory effects
|
Follicular lymphoma, leukemia, nasopharyngeal carcinoma
|
Upregulation of BiP and CHOP mRNA; increased protein levels of BiP, IRE1, p-PERK, CHOP, p-eIF2α and XBP1; induced caspase-3 and PARP cleavage
|
||
|
Rhein
|
Natural product
|
Preclinical
|
Antibacterial agent
|
Nasopharyngeal carcinoma
|
Increased BiP expression, upregulated PERK and ATF6 pathways, increased ROS
|
||
|
Sarsasapogenin
|
Natural product
|
Preclinical
|
(ROS)-mediated mitochondrial dysfunction, medicinal Chinese herb
|
Cervical cancer
|
Upregulated CHOP expression, upregulated cytochrome c and caspase activation, induced ROS
|
Shen et al. [165]
|
|
|
Sorafenib (Nexavar)
|
Small molecule
|
Phase I/II/III/IV
|
2013
|
Multiple kinase inhibitor (cytotoxic)
|
AML, breast cancer, hepatocellular carcinoma, HNSCC, melanoma, non-small cell lung cancer, pancreatic cancer, renal cell carcinoma, soft tissue sarcoma
|
Induced splicing of XBP1 mRNA, increased CHOP and GADD34 gene expression, induced phosphorylation of eIF2α
|
Yi et al. [166]
|
|
Vemurafenib (Zelboraf)
|
Small molecule
|
Phase I/II/III/IV
|
2011
|
BRAF(V600E) kinase inhibitor
|
Advanced solid tumors, colorectal cancer, CR prostate cancer, hairy cell leukemia, mutant/metastatic melanoma
|
Sequestration of Bip, PERK activation, increased XBP1 splicing, increased phosphorylation of eIF2α, induced gene expression of ATF4, CHOP, ATF3
|
Beck et al. [167]
|
The degree to which these compounds directly activate the UPR, or whether activation is a late effect of the reduced energetics of a dying cell, will continue to be elucidated. The idea that an overwhelming ER stress might tilt the balance of the two arms of the UPR from adaptation toward apoptosis as a chemotherapeutic strategy is well characterized in these studies. The question with regard to activation vs. inhibition of the UPR, as a more productive approach to reducing tumor burden, hinges on whether inhibition can be therapeutically maintained or if the applied stress can be selectively activated to kill only malignant cells. The answer is not as simple as trying to align the plethora of data about individual UPR players and the compounds that lead to their accumulation. The fact that the three branches of the UPR (Fig. 10.1) are in many cases redundant (e.g., CHOP can be upregulated by PERK-ATF4 signaling or ATF6 activation, and all three arms of the UPR can increase cytoprotective chaperones, including GRP78/BiP) creates an onerous burden for the specific small molecule inhibitor approach. An effective UPR-inhibiting anticancer approach would therefore need to inhibit all three ER stress axes to be effective. Although UPR inhibition might reduce angiogenesis and perhaps invasion, even with constant applied pressure, a likely outcome is smoldering disease.
The UPR is a promising therapeutic target for many cancers, including HNSCC [101]. The underlying hypothesis for targeting the UPR is based largely on the highly secretory nature of malignant cells. The drive for vascularization of the stroma relies heavily on tremendously increased levels of protein turnover and constitutively upregulated expression of protein-folding machinery [168, 169]. The paradox of activation versus inhibition of the UPR in tumor development was addressed in a study by Mimura et al. in which it was demonstrated that a synergy existed between MKC-3946, a small molecule inhibitor of the IRE1α endoribonuclease and UPR-inducing bortezomib [170]. MKC-3946 was able to simultaneously “step on the gas” of the apoptotic arm of the UPR while “releasing the brake” of the adaptive arm. Interestingly, GRP78/BiP expression, whether increased or decreased, has been shown to improve anticancer therapies in several studies. GRP78 knockdown interfered with AKT signaling and enhanced cisplatin-mediated cell killing [171], decreased DNA repair capacity, and reduced survival in virally transformed fibroblasts [172]. On the other hand, ER stress and increased BiP expression primed Chinese hamster ovary and colon cancer cells to melphalan, 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) and cisplatin [173, 174]. The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) induced ER stress and sensitized oral cancer cells to cisplatin [175]. It is possible that the general pause in translation that occurs during ER stress prevents the de novo synthesis of DNA synthesis and repair enzymes (e.g., ERCC1, XRCC1, and MSH2–6) required to combat the effects of cisplatin and other DNA-damaging agents. We support the continued effort to discover novel compounds that increase UPR signaling to cytotoxic levels in malignant cells predicated on the facts that (1) many cytotoxic FDA-approved anticancer drugs increase, not decrease, the UPR; (2) UPR inhibition as an anticancer approach requires constant applied pressure to all three arms of the UPR and might lead to smoldering disease; (3) enhanced UPR activation primes cancer cells for death in response to standard of care drugs; and (4) a systemic UPR challenge might be delivered to selectively kill cells with increased UPR (i.e., malignant cells), whereas adjacent healthy cells might mount an effective adaptive UPR and return to homeostasis.
10.3 High-Throughput Screening to Identify Novel UPR Activators
The last decade has seen tremendous advances in the technology and methods required for high-throughput screening (HTS) and drug discovery. The development of biochemical and cell-based assays to rapidly discover bioactive small molecules and natural products in high-density formats has been the focus of both industry and academic laboratories all over the world [176]. Sophisticated confirmatory secondary and tertiary in vitro assays and cutting edge animal models have been developed to validate hit compounds. Drug discovery teams comprised of biologists, medicinal, computational, and synthetic chemists and bio-informatics personnel use these model systems to guide their efforts to develop hit compounds into a more potent and specific lead compound [176–179]. Access to novel academia-friendly integrated compound management software platforms has provided a necessary functional link between enormous data sets and the identification of hit and lead compounds [180]. The application of informatics sciences is an essential component of drug discovery at two critical junctures: (1) HTS data management, which often requires the ability to search/parse hundreds of thousands to millions of data points, and (2) analytic structure analysis for hierarchical clustering to identify novel hit chemotypes and useful (druggable) chemical signatures. A wide variety of HTS cell-based assays have been and are currently utilized in the search for the next family of anticancer compounds.
Predicated on the discovery that the proteasome inhibitor bortezomib induced ER stress and UPR-dependent apoptosis in HNSCC, we sought to develop an HTS platform to screen libraries of small molecules and natural products to find more potent and selective UPR modulators [22]. An ~66,000 compound library that included a collection of 5036 natural product extracts at the University of Michigan Center for Chemical Genomics and the US National Cancer Institute (NCI) collection of approximately 350,000 compounds in collaboration with the Conrad Prebys Center for Chemical Genomics at the Sanford Burnham Medical Research Institute were screened. Briefly, our screening strategy utilized a complementary cell-based reporter assay with two CHO-K1 cell lines individually transfected with luciferase reporters to monitor Chop promoter activation or Xbp1
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


