Trial name
Compound
Clinical trial number
Reirradiation With MK-3475 in Locoregional Inoperable Recurrence or Second Primary Squamous Cell CA of the Head and Neck
Pembrolizumab
NCT02289209
Study of MK-3475 (Pembrolizumab) in Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma After Treatment With Platinum-based and Cetuximab Therapy (MK-3475-055/KEYNOTE-055)
Pembrolizumab
NCT02255097
A Combination Clinical Study of PLX3397 and Pembrolizumab To Treat Advanced Melanoma and Other Solid Tumors
PLX3397, Pembrolizumab
NCT02452424
Pembrolizumab (MK-3475) Versus Standard Treatment for Recurrent or Metastatic Head and Neck Cancer (MK-3475-040/KEYNOTE-040)
Pembrolizumab
NCT02252042
A Study of Pembrolizumab (MK-3475) for First Line Treatment of Recurrent or Metastatic Squamous Cell Cancer of the Head and Neck (MK-3475-048/KEYNOTE-048)
Pembrolizumab
NCT02358031
Trial of Nivolumab vs Therapy of Investigator’s Choice in Recurrent or Metastatic Head and Neck Carcinoma (CheckMate 141)
Nivolumab
NCT02105636
A Phase I Study of an Anti-KIR Antibody in Combination With an Anti-PD1 Antibody in Patients With Advanced Solid Tumors
Lirilumab, nivolumab
NCT01714739
A Study of the Safety, Tolerability, and Efficacy of INCB24360 Administered in Combination With Nivolumab in Select Advanced Cancers
Nivolumab, INCB24360
NCT02327078
A Study to Investigate the Safety and Efficacy of Nivolumab in Virus-associated Tumors (CheckMate358)
Nivolumab
NCT02488759
Phase II Study of MEDI4736 Monotherapy in Treatment of Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck
MEDI4736
NCT02207530
Study of MEDI4736 Monotherapy and in Combination With Tremelimumab Versus Standard of Care Therapy in Patients With Head and Neck Cancer
MEDI4736, tremelimumab
NCT02369874
Study to Assess Combination of MEDI4736 With Either AZD9150 or AZD5069 in Patients With Metastatic Squamous Cell Carcinoma of Head and Neck
MEDI4736, AZD9150, AZD5069
NCT02499328
A Phase 1 Study of MPDL3280A (an Engineered Anti-PDL1 Antibody) in Patients With Locally Advanced or Metastatic Solid Tumors
MPDL3280A
NCT01375842
A Phase I, Open-Label, Multicentre Study to Evaluate the Safety, Tolerability and Pharmacokinetics of MEDI4736 in Patients With Advanced Solid Tumors
MEDI4736
NCT01938612
PD-L1
Programmed death receptor ligand-1 (PD-L1) is the ligand for PD-1, and ligation of-PD-L1 to PD-1 leads to T cell inhibition [49]. PD-L1 is expressed in a multitude of tissues including muscles and nerves. Of relevance for cancer immunotherapy, PD-L1 can be expressed on the surface of tumor cells, tumor-associated macrophages (TAMs), and T lymphocytes and can subsequently inhibit PD-1-positive T cells [48, 49]. The expression of PD-L1 can be induced by cytokines such as interferons, or alternatively PD-L1 can be expressed autonomously through aberrations in the EGFR signaling pathway [50–53]. In clinical studies, antibodies blocking the PD-L1/PD-1 interactions have demonstrated 6–21 % overall response rate (ORR) in nonselected tumors [54, 55], 19 % in triple-negative breast cancer [56], and 38 % in patients with NSCLC demonstrating high PD-L1 expression [57]. The toxicity profile for anti-PD-L1, like anti-PD-1, is thought to be distinct and generally milder compared to the adverse events observed with anti-CTLA-4 [55]. The mechanism of action of anti-PD-L1 is currently under investigation. However, several studies have shown that clinical benefit is directly correlated with high expression of PD-L1 [57]. The multiple open clinical trials testing the efficacy of anti-PD-L1 in HNSCC are summarized in Table 9.1.
Other checkpoint blockade agents
A large number of immune checkpoint inhibitors have been discovered, and a significant portion of them are currently being tested in either a preclinical setting or directly in clinical trials for the treatment of cancer. Two examples that have moved into the clinical setting are anti-LAG-3, targeting an inhibitory receptor on the surface of T cells, and anti-KIR, to reverse the inhibited natural killer (NK) cells. Trials involving anti-LAG-3 and anti-KIR are ongoing in the treatment of hematologic and advanced solid malignancies.
9.3 Adoptive T-Cell Therapy with Autologous T Cells
Regardless of HPV status, the presence of tumor-infiltrating lymphocytes is associated with more favorable outcomes in HNSCC [6, 7]. Studies in other types of cancers, most notably colon cancer, have followed a similar pattern: patients with high CD3 and CD8 infiltrate are associated with significantly better course of disease than those with low T-cell infiltrates [58].
Adoptive T-cell therapy (ACT) involves harvesting T cells from autologous tumor resection or biopsy, expanding them ex vivo with IL-2, testing for tumor specificity, and then rapidly expanding them for reinfusion into a non-myeloablative lympho-depleted patient. High-dose IL-2 is also administered to support the in vivo expansion of transferred T cells [59, 60]. CD8 T cells often constitute the majority of the tumor-specific T cells in culture; other cells such as CD4 T cells, NK cells, and gamma-delta T cells are also present in culture and have been shown to possess significant antitumor activity [60]. Regulatory T cells are also present in the culture, and depletion of them prior to reinfusion positively correlated with increased response to ACT [61–64].
In metastatic melanoma, tumor-specific T cells can be recovered from 35 to 70 % of patients, and the response in these patients is 50–75 % [65–67]. Notably, 95 % of the complete responders demonstrated a durable response for at least 5 years [67]. The use of ACT for patients with HNSCC has been pioneered at a few centers, with one reporting a remarkable response rate of 43 % in the seven patients studied [68]. Of note, a significant hurdle to adoptive T-cell therapy is the ability to culture and expand tumor-specific T cells from the patients’ autologous tumors. In renal, breast, and colon cancer, the recovery rate is extremely low (0–20 % [69]). Interestingly, in HNSCC the success in obtaining tumor-specific T cells from autologous tumors is around 60 %, a number similar to melanoma [70, 71]. In our center, we have successfully cultured tumor-specific T cells from a similar percentage of patients and have treated a single patient with HNSCC.
Strategies to increase the success of recovering tumor-specific T cells from tumor specimen are currently being studied. Data from our group generated using multispectral fluorescent IHC suggest that the ratio of CD8 effector T cells to FoxP3+ Treg is an important biomarker that predicts the success of culturing tumor-specific T cells in melanoma (Feng et al, Manuscript in revision). Our group is applying the same multispectral imaging strategy to characterize the tumor environment in HNSCC (Fig. 9.1). Modulation of the tumor environment either during culture or prior to surgery can potentially enhance this ratio, leading to greater recovery of tumor-specific T cells. Administration of 4-1BB agonist antibody during culture has been shown in patients with melanoma to increase the tumor-specific T-cell function [72], and a study evaluating the effect of neoadjuvant anti-PD-1 therapy on success of culturing tumor-specific T cells is underway.
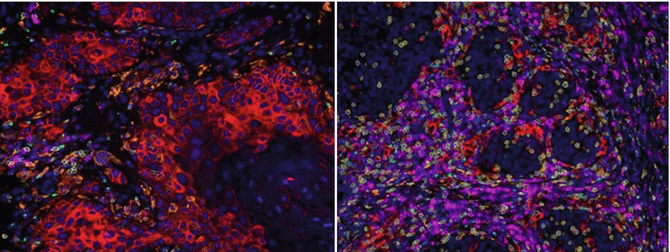
Fig. 9.1
Distinct tumor microenvironment in HNSCC. Left: Tumor with low immune infiltrate and high PD-L1 expression. Right: Tumor with high immune infiltrate and low PD-L1 expression. Key – Red: PD-L1; Yellow: CD8; Green: FoxP3; Magenta: CD79A (B cells); Orange: CD163 (Macrophages); Blue: DAPI
9.4 Cytokine-Based Therapy
Cytokines are molecular signaling molecules that allow intercellular communication over a long distance to generate rapid and robust immune responses in a controlled manner. The immune-mediated antitumor activities of a few cytokines have been well characterized in preclinical settings and led to many clinical trials testing the efficacy of interleukins including IL-2, IL-7, IL-12, IL-15, and IL-21, interferons, and GM-CSF. Out of these, high-dose IL-2 was approved for the treatment of metastatic melanoma and renal cell carcinoma [73], and IFN-α was approved for adjuvant therapy for stage 3 melanoma [74].
Interleukin-2 (IL-2) is a member of the IL-2-related family that functions to stimulate T-cell growth through the IL-2 receptor. IL-2 exhibits myriad effects on the immune system in addition to promoting CD4 and CD8 T-cell proliferation and differentiation into effector T cells and memory T cells. IL-2 also expands Tregs, which express high-affinity IL-2 receptors [75, 76]. This acts as a negative feedback mechanism to decrease CD8 T-cell activity through both depletion of IL-2 and inhibition by Tregs.
Systemic high-dose IL-2 plays a key role in the treatment of metastatic melanoma and renal cell carcinoma. Although limited to patients in otherwise good health due to acute toxicities, high-dose IL-2 induces objective clinical response in 15–25 % of patients with metastatic melanoma and renal cell carcinoma, with complete response around 7–12 % [77–80]. Importantly, of the patients achieving complete response, 70–90 % remains disease-free in an ongoing 20-year follow-up [78, 81].
The use of IL-2 in HNSCC has been limited to local-regional or intralesional injection with mixed results. High-dose IL-2 injected local-regionally demonstrated induction of tumor-specific T cells in the draining lymph nodes of patients with HNSCC [82]. However, perilymphatic injection of low-dose, but not high-dose, IL-2 demonstrated temporary regression of HNSCC and two complete responders in lip cancer [83–87]. The mechanism for resistance to IL-2 therapy in HNSCC is unclear. It has been suggested in melanoma that patients who fail to demonstrate significant Treg induction experienced an increased response to therapy [88]. One explanation for these observations is that the presence of abundant regulatory T cells unique to HNSCC could be rapidly expanded with IL-2 and limit the antitumor CD8 response. Combination therapy with systemic high-dose IL-2 and Treg depleting agents, such as cyclophosphamide, may be an avenue to overcome this pathway for the treatment of HNSCC.
Interferon-α (IFN-α): IFN-α is a type I interferon that is involved in the immune response to viral infections. In cancer immunotherapy, administration of IFN-α can activate CD8 T cells as well as the innate immune system, specifically NK cells, DCs, and macrophages [89, 90]. This has led to a heightened antitumor immune response in preclinical models, especially when used in combination therapy with checkpoint blockades [29]. The use of IFN-α clinically has so far been limited to adjuvant setting in high-risk melanoma where it significantly improves disease-free survival; however, the overall survival benefit seems to be limited to a very small subset of patients [74]. Currently IFN-α is being tested in solid tumors in combination with various targeted and immunotherapy agents.
Other cytokines
Administration of cytokines such as IL-7, IL-12, IL-15, IL-21, and GM-CSF and inhibition of suppressive cytokines such as TGF-β are still being explored in the treatment of a variety of solid malignancies, including HNSCC. IL-12 has been reported to increase B-cell distribution and activation when injected intratumorally in an adjuvant setting for the treatment of HNSCC; however, the clinical benefit is currently unclear [91, 92]. It is possible that we may see an increasing number of these agents used in combination therapies for HNSCC, as preclinical evidence points to increased efficacy of these agents when used in combination with checkpoint blockade or T-cell agonists.
9.5 Immune System Agonists
Overview
Upon encountering an antigen, T cells can upregulate co-stimulatory receptors such as OX40 and 4-1BB [24]. Activation of these co-stimulatory receptors can significantly increase T-cell proliferation and function, leading to a greater antitumor immune response. A few of these co-stimulatory receptors with clinical application potential are discussed below.
OX40
OX40 is a member of the tumor necrosis factor receptor (TNFR) superfamily and is present on surface of T cells, in particular CD4 T cells and Tregs [63]. Activation of OX40 through an agonist antibody, either directly or indirectly, increases CD4 and CD8 T-cell priming, proliferation, and function [63]. Interestingly, while OX40 activation in Tregs does not seem to affect their proliferation, their ability to inhibit the function of CD8 T cells appears to be hindered partly through the disruption of FoxP3 expression and inhibitory cytokine release [62, 93]. This tips the effector to inhibitor balance in the tumor microenvironment and results in significant antitumor efficacy in many immunogenic preclinical models [94]. In poorly immunogenic models, combination with a vaccine appears to substantially improve therapeutic efficacy (unpublished). Clinically, OX86, an agonist antibody to OX40, has demonstrated great safety in a phase I trial [95], and the antibody is currently being tested as a single agent in neoadjuvant setting for the treatment of patients with HNSCC (NCT02274155). Based on preclinical evidence, OX40 antitumor activity could potentially be further improved by combining it with a vaccine.
4-1BB
4-1BB is another member of the TNFR superfamily expressed stably on activated T cells and NK cells [96]. Its activation by 4-1BBL or by an agonist antibody on CD8 T cells results in increased proliferation, cytokine production, and survival [97]. 4-1BB activation also has profound impact on the humoral immune system and CD4 T cells [98]. Studies in preclinical models of lupus demonstrated suppressed B-cell response and induction of CD4 T-cell anergy upon 4-1BB stimulation [99]. In preclinical tumor models, in vivo administration of 4-1BB agonist antibody demonstrated enhanced antitumor activity in a CD8 T-cell-dependent manner [100, 101], and currently these agents are being tested in clinical trials for the treatment of solid and hematologic malignancies. 4-1BB agonists, either as a single agent or in combination with checkpoint blockade, are currently being proposed for the treatment of HNSCC, and new trials could open in the near future.
CD40
CD40 is another member of the TNFR superfamily that is mainly expressed on the surface of antigen-presenting cells (APC). CD40 plays an important role in the licensing of APCs by T cells. Ligation of CD40 to CD40L or an agonist antibody leads to expression of major histocompatibility complexes (MHC) and co-stimulatory molecules, secretion of proinflammatory cytokines (e.g., IL-12), and increased antigen-presenting capacity. This in turn facilitates the priming of CD4 and CD8 T cells, leading to the activation of NK cells and to enhance antitumor immune response. CD40 agonist antibodies such as CP-870,893 (Pfizer) and dacetuzumab (Seattle Genetics, discontinued) demonstrated preclinical therapeutic efficacy in both solid and hematologic malignancies. Among solid tumors, CD40 agonist antibodies had been tested in the treatment of pancreatic cancer with objective response rate of around 20 %. A preliminary study with combination CP-870,893 and tremelimumab (anti-CTLA-4, Pfizer) was performed with an ORR at 27.3 % and CR at 9.1 % [102]. The utilization of anti-CD40 in HNSCC has not yet been extensively explored.
9.6 Vaccines
Overview
A coordinated antitumor immune response that induces both CD4 and CD8 T cell is critical to mediating therapeutic effect. To activate tumor-specific CD4 and CD8 T cells, tumor-associated antigens (TAAs) are processed and cross-presented by APCs. When the immune system is functioning optimally and in the presence of adequate co-stimulatory signals and cytokines, T cells are activated and expanded in numbers that can recognize and eliminate tumor cells. It is currently hypothesized that a preexisting antitumor T-cell response is critical for the therapeutic effects of checkpoint blockade, especially anti-PD-1. For patients lacking this preexisting tumor-specific immune response, priming a new antitumor response through vaccines is expected to provide substantial benefit to therapy containing checkpoint blockade or immune system agonists. The many forms of vaccines will be briefly discussed below.
Peptide and whole protein vaccine
The utilization of peptide vaccine for the treatment of cancer has expanded since one of the first studies used a MAGE-1 peptide for the treatment of melanoma [103]. Delivery of peptide-based vaccine with an immune adjuvant or pulsed onto DCs has been shown to elicit peptide-specific CD8 T-cell responses. Peptide cocktails have been used with various degrees of success in certain solid tumors, with one study using IDM-2101, a 10-peptide vaccine cocktail for the treatment of patients with stage IIIB and IV NSCLCs. The study reported no significant adverse effect with 17.3-month median overall survival [104]. For vaccines containing single whole protein, progression-free survival and median overall survival benefit have also been observed [105, 106]. These results led to the first FDA-approved cancer vaccine, Sipuleucel-T, for the treatment of castration-resistant prostate cancer. However, no durable responses were observed with single peptide vaccines, potentially owing to immune evasion as a result of protein downregulation by tumor cells [107].
Whole-cell vaccines
Whole-cell vaccines are prepared using irradiated whole tumor cells and are frequently delivered with an adjuvant, such as GM-CSF. One advantage of whole-cell vaccines, compared to a peptide vaccine, is that it contains a richer antigenic source. By using the autologous tumor, multiple relevant antigens can potentially be targeted in a single vaccine, aimed to induce antitumor immune response when combined with a stimulatory adjuvant. Genetic modification of the tumor has also been accomplished, particularly with GM-CSF, which stimulates recruitment of dendritic cells (DC) to the vaccine site and results in augmented antigen processing and presentation to both T and B cells [108]. Due to the cost and difficulty of coordinating autologous vaccine preparation, allogeneic vaccine strategies now represent the bulk of whole tumor vaccine trials [109]. The assumption with allogeneic vaccines is that many antigens are shared between tumors so that vaccination with an allogeneic tumor can effectively prime an immune response toward antigens commonly overexpressed by a patient’s autologous tumor. This approach demonstrated promise with one phase II study reporting median survival of 24.8 months in patients with pancreatic adenocarcinoma vaccinated with a GM-CSF secreting allogeneic tumor [110].
Autophagosome-based vaccines
Autophagy, the process by which cells recycle cellular components through autophagolysosomal fusion, is essential in the immune recognition of cancer [111–113]. Tumor autophagy is necessary for tumor-specific T-cell priming through the induction of cross-presentation of tumor antigens by DCs [114, 115]. In addition, tumor-derived autophagosomes contain short-lived proteins and defective ribosomal products, which represent the major source of proteins for MHC class I-restricted self-peptides [116, 117]. These two types of proteins degrade rapidly and are not enriched in whole-cell vaccines but are captured and enriched in the autophagosome-based vaccines. Antitumor efficacy has been shown in several different preclinical models [114, 118, 119], and DRibbles (UbiVac), an autophagosome-based vaccine, is currently being tested in an ongoing multicenter phase II clinical trial in patients with non-small cell lung cancer (NCT01909752). A trial for the treatment of patients with HNSCC is currently in the planning stages.
HPV vaccine
HPV-related HNSCC has been steadily on the rise for the last three decades in the United States with a concurrent decrease in smoking-related HNSCC [120]. It is estimated that by 2020, HPV-related HNSCC cases will be more frequent than HPV-negative cases [121]. HPV is a DNA virus that can inhibit the function of P53 and Rb through the E6 and E7 oncoproteins, respectively. A direct causal relationship has been established between high-risk HPV and HNSCC, and the E6 and E7 oncoproteins can be readily detected in many HPV-positive tumors. The therapeutic use of HPV vaccine is still being explored in HNSCC; studies in patients with cervical intraepithelial neoplasia grade 2/3 demonstrated that intramuscular vaccination with HPV E6/E7 vaccine induced CD8 T-cell response that localized to the dysplastic mucosa. Currently, there are multiple trials opened for the treatment of HNSCC using HPV vaccines delivered using different platforms. These include ADXS 11-011 (Advaxis Inc.) utilizing Listeria as a vector for vaccine delivery, (NCT 02002182) using peripheral blood mononuclear cells pulsed with E6 and E7 peptide, and (NCT00019110) employing intramuscular vaccination with VGX3100 and INO-9012 (Inovio Pharmaceuticals) followed by electroporation (NCT02163057). Additionally, a vaccine against high-risk HPV subtypes has successfully reduced the incidence of cervical intraepithelial neoplasia (CIN) by 90–96 % [122, 123] and is currently being implemented in the prevention of HPV-induced HNSCC (NCT02382900).
9.7 Biomarkers
The final part of this chapter will cover in brief biomarkers, a rapidly advancing area for immunotherapy. With increasing numbers of immunotherapy strategies available for the treatment of solid malignancies, including HNSCC, identification of prognostic and predictive biomarkers is paramount to stratify patients and direct therapy. Understanding these biomarkers is an active area of research, with a majority of the attention focusing on the checkpoint blockades such as anti-PD-1. Recent studies have demonstrated that response to anti-PD-1 is heavily dependent on the expression of PD-L1 in the tumor microenvironment as well as the mutational burden of the tumor itself [48, 124, 125]. These findings suggest that the response to anti-PD-1 is hinged on a preexisting antitumor immune response and that anti-PD-1 acts to free the CD8 T cells from inhibition to exert their antitumor activities. A biomarker for anti-CTLA-4 is also being researched; preliminary studies suggest that the response is dependent on FcgRIIIA (CD16)-expressing monocyte-mediated elimination of Tregs [32]. IHC is the most frequently used technique in the identification of these biomarkers. Studies using quantitative IHC have identified CD8 T-cell infiltration as an important prognostic factor in predicting outcome for patients with HNSCC [6, 7]. More recently, multiplex immunohistochemistry has emerged as a more advanced tool for the analysis of the tumor microenvironment, offering a more efficient and comprehensive analysis of the tumor microenvironment for both diagnostic and mechanistic studies [126–128]. This also allows us to explore the relationship between cell types in the peritumoral and intratumoral compartments, quantify the expression of specific biomarkers (Fig. 9.1), and examine their impact on patient prognosis and response to therapy. As research on biomarkers advances, we will hopefully be able to better direct patients, based on their tumor microenvironment and mutational status, to the immunotherapy that potentially offers the best response rate and ultimately a cure for their disease.
References
1.
Mehra R, Cohen RB, Burtness BA. The role of cetuximab for the treatment of squamous cell carcinoma of the head and neck. Clin Adv Hematol Oncol. 2008;6(10):742–50.PubMedCentralPubMed
2.
Argiris A, et al. Head and neck cancer. Lancet. 2008;371(9625):1695–709.PubMed
3.
Wong RJ, Shah JP. The role of the head and neck surgeon in contemporary multidisciplinary treatment programs for advanced head and neck cancer. Curr Opin Otolaryngol Head Neck Surg. 2010;18(2):79–82.PubMed
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


