Mammary Analogue Secretory Carcinoma
Polymorphous Low-Grade Adenocarcinoma
Epithelial–Myoepithelial Carcinoma
Carcinoma Ex Pleomorphic Adenoma
Introduction
Although the etiology of salivary gland tumors is unknown, the involvement of environmental and genetic factors has been suggested. Over the years, there has been some progress in clarifying specific causes of salivary gland cancer, and several risk factors have been identified. Compared with most other head and neck cancers, malignant salivary gland tumors have not been associated with smoking or excess alcohol intake. The possible risk factors include radiation exposure, hormones, exposure to several occupational risk factors, viral infections, and genetic predisposition. The best-known risk factor is radiation exposure, as evident in atomic bomb survivors and in patients receiving therapeutic radiation.
Recent studies have clearly shown that the development and progression of human malignancies is associated with an accumulation of alterations in protooncogenes and tumor-suppressor genes (TSGs), and this also appears to be the case with salivary gland tumors. The pathogenesis of salivary gland neoplasms is a heterogenous process involving several pathways, and it has been proposed that the tumor genotype affects the clinical behavior and prognosis. Obviously, there are abnormalities in the genome that underlie salivary gland neoplasms, but little information has so far been reported about specific genetic and epigenetic alterations in salivary gland carcinomas. With the advent of gene and protein expression profiling, future molecular and genetic insights will enhance our understanding of salivary gland carcinogenesis.
Radiation
The evidence that exposure of salivary gland tissue to ionizing radiation increases the risk of developing salivary tumors is considerable. Long-term follow-up studies of survivors of the atomic bomb explosions in Hiroshima and Nagasaki show an increased relative risk of 3.5 for benign neoplasms and 11 for malignant salivary neoplasms.1 The risk was directly related to the level of exposure to ionizing radiation. In particular, there was a higher frequency of mucoepidermoid carcinoma, pleomorphic adenomas, and other salivary carcinomas.2
Therapeutic radiation has also been linked with an increased risk of salivary gland tumors in comparison with an unexposed control group.3 Patients receiving iodine 131 in the treatment of thyroid disease are at increased risk for salivary gland carcinoma, as the isotope is also concentrated in the salivary glands.4 Excessive use of medical and dental diagnostic radiography may also play a role in triggering salivary gland tumors.5 An increased occurrence of salivary malignancies in children with acute leukemias treated with multiagent chemotherapy and prophylactic cranial irradiation has also been noted.6
However, there appears to be no excess risk in those exposed to radon,7 microwaves from cellular phones,8 or increased exposure to ultraviolet radiation.9
Viruses
Among potential viral etiologies, only Epstein–Barr virus infection has been implicated in the pathogenesis of salivary lymphoepithelial carcinomas, which are more common in the Inuit and Chinese than in Western populations.10 Epstein–Barr viral genomes can be detected in the malignant cells.11 There is a marked histological similarity to undifferentiated nasopharyngeal carcinoma, which has also been linked with Epstein–Barr virus.
However, no increased risk has been documented for infections with herpesvirus, human papillomavirus, human immunodeficiency virus (HIV), or other viruses.
 With the exception of salivary lymphoepithelial carcinoma, viral infections are not associated with salivary gland cancer.
With the exception of salivary lymphoepithelial carcinoma, viral infections are not associated with salivary gland cancer.
Lifestyle and Nutrition
Unlike other head and neck cancers, alcohol and smoking abuse are not associated with an increased risk of salivary cancers,12 with the exception of the substantially increased risk evident from the association of smoking with benign Warthin tumor (see Chapter 19). Exposure to silica dust and kerosene as a cooking fluid have been found to increase the risk of salivary malignancies in a Chinese population.13 An increased level of risk has been postulated in patients with high cholesterol levels.14
 At present, lifestyle is not known to have any influence on the development of salivary gland cancer.
At present, lifestyle is not known to have any influence on the development of salivary gland cancer.
Occupation
Occupational exposures that have been reported to be associated with an increased risk of salivary carcinoma include asbestos mining, manufacturing of rubber, exposure to metal in the plumbing industry, and some woodworking occupations.15 There has also been a study showing a high relative risk of developing salivary carcinoma in workers with exposure to nickel, chromium, asbestos, and cement dusts.16 One interesting finding is an elevated risk of salivary gland cancer in women employed as hair-dressers and those working in beauty salons.17 However, this observation has never been confirmed in other studies.
 The evidence that specific types of occupational exposure are associated with an increased risk of salivary gland cancer is low.
The evidence that specific types of occupational exposure are associated with an increased risk of salivary gland cancer is low.
Hormones
The expression of endogenous hormones in normal and neoplastic salivary glands is a controversial topic. Estrogen receptors (ERs) have been found in normal salivary glands in females and males, but the studies have shown conflicting results.18 ERs were reported in some cases of acinic cell and mucoepidermoid carcinoma, but not in adenoid cystic carcinoma.19 ERs have been reported in pleomorphic adenoma in some studies,19 but not in others.20 Similarly, progesterone receptors (PRs) have been reported in normal salivary glands and in tumors.21 High levels of PR were demonstrated in recurrent pleomorphic adenoma,21 and this was thought to be a prognostic factor.
In conclusion, the incidence of ER and PR in salivary gland cancer is variable and it probably does not play any important role in the development of salivary gland tumors. Hormone receptor expression is virtually absent in most cases of salivary cancer, with the exception of androgen receptors (ARs) in salivary duct carcinomas.22 One recent study showed immunoreactivity for AR in all cases of salivary duct carcinoma and in most cases of carcinoma ex pleomorphic adenoma,20 suggesting that the patients might benefit from antiandrogen therapy.23
Genetics
Despite advances, the genetic events associated with the development and progression of salivary gland neoplasia are still largely unknown. There is a clear need for a better understanding of such events in order to define new prognostic and diagnostic markers and design targeted therapeutic interventions.
The recent application of expression microarray technologies in the study of salivary gland cancer has led to interesting new data.24–27 Studies of microsatellite and single-nucleotide polymorphism (SNP) markers and comparative genomic hybridization (CGH) arrays have identified losses of heterozygosity (LOH) and various chromosomal aberrations in various types of salivary gland carcinoma.28–30 Analysis of methylation patterns has also been performed to detect epigenetic alterations in several salivary gland tumors.31,32 These methods together have identified a batch of chromosomal areas or genes that participate in the development and progression of different types of salivary gland tumor.
Like benign tumors, malignant salivary gland tumors are also frequently characterized by recurrent chromosome translocations. In addition, a substantial proportion of sporadic translocations have been discovered in salivary gland tumors.33,34 As in benign lesions, PLAG1, HMGA2, MECT1, and MAML2 rearrangement are found. In addition, several other recurrent or sporadic translocations—involving, for example, tyrosine kinase NTRK3, transcription factors ETV6, MYB, and NFIB, and several others, are found in salivary gland tumors. As mentioned above, the translocated genes encode novel fusion proteins as well as abnormally expressed normal or altered proteins activating different signaling pathways that are involved in the origin and progression of tumors.34–36
 Acinic Cell Carcinoma
Acinic Cell Carcinoma
Acinic cell carcinoma (ICD–O code 8550/3) generally shows few genetic alterations. In the most extensive study, El-Naggar et al.37 reported the highest incidence of LOH, suggesting the presence of tumor suppressor gene (TSG) at chromosomes 4p15–16, 6p25-qter, and 17p11. LOH was significantly associated only with tumor grade. No apparent correlation between LOH and other clinicopatho-logical characteristics was identified.
Acinic cell carcinomas with high-grade transformation are composed of an ordinary acinic cell carcinoma and a poorly differentiated high-grade component, in variable proportions. Transformed high-grade acinic cell carcinomas are associated with a poor clinical outcome, as they tend to recur and show perineural and angiolymphatic intravascular invasion.38 The clinical course in most cases is fatal, with dissemination and tumor-related death in most patients. Although high-grade transformation is always associated with tumor progression, little is known about the molecular-genetic events that regulate it. Alteration of the p53 pathway has been shown to play a critical role in the tumorigenesis of many human malignancies, but not in acinic cell carcinoma.38 KIT, ERBB2 (formerly HER–2/neu), and TP53 do not appear to play a significant role in the process of high-grade transformation of acinic cell carcinomas. A role of cyclin D1 in high-grade transformation of acinic cell carcinomas has been postulated, and molecular analysis of the cyclin D1 gene is therefore needed.38
 Mammary Analogue Secretory Carcinoma
Mammary Analogue Secretory Carcinoma
Mammary analogue secretory carcinoma (MASC) is an unusual, hitherto undescribed, but distinctive salivary gland tumor characterized by strong vimentin and S100 protein positivity, with strong histomorphological and immunohistochemical resemblance to secretory carcinoma of the breast, and histomorphological features shared with conventional salivary acinic cell carcinomas.36
MASCs of the salivary glands, like secretory carcinoma of the breast, have been shown to harbor a recurrent balanced chromosomal translocation, t (12;15)(p13;q25), which leads to a fusion gene between the ETV6 gene on chromosome 12 and the NTRK3 gene on chromosome 15 (Fig. 26.1). The biological consequence of the translocation is fusion of the transcriptional regulator (ETV6) with membrane receptor kinase (NTRK3), which activates cell proliferation and survival. The presence of the ETV6-NTRK3 fusion gene has not been demonstrated in any other salivary gland tumor so far, and it therefore appears that MASC and salivary acinic cell carcinoma are distinct entities and should be recorded separately in classifications of salivary gland tumor.36
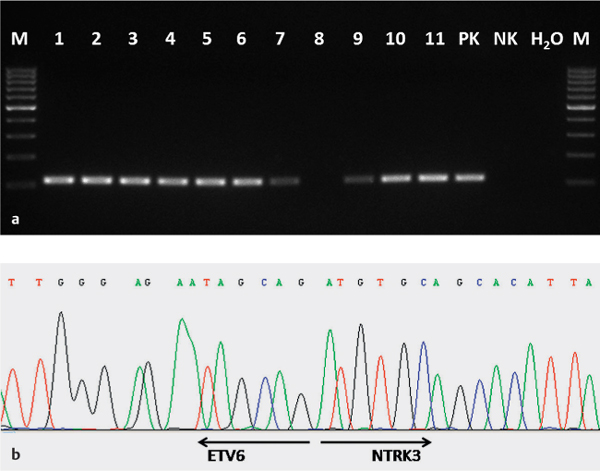
Fig. 26.1 a, b The ETV6-NTRK3 fusion gene in a mammary analogue secretory carcinoma.
a Expression of the ETV6-NTRK3 fusion transcript in the mammary analogue secretory carcinoma using reverse transcriptase polymerase chain reaction (RT–PCR). 1–11, samples from the mammary analogue secretory carcinoma (all except no. 8 are positive); PK, positive amplification control; NK, negative amplification control; H2O, water.
b Sequence analysis of ETV6-NTRK3 gene fusion. The arrows show translocation breakpoints.
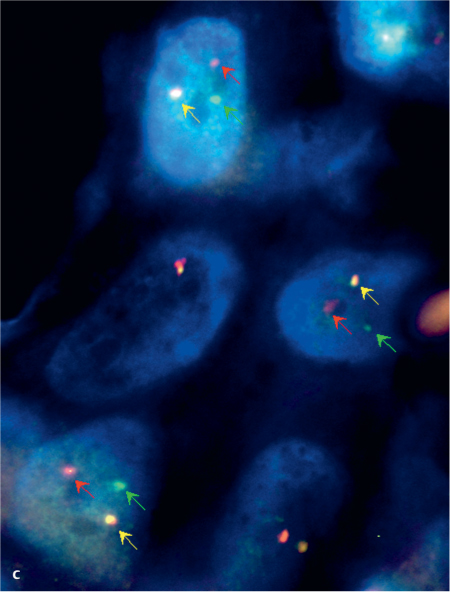
Fig. 26.1 c Fluorescence in situ hybridization analysis of a mammary analogue secretory carcinoma using an LSI ETV6 (TEL) (12p13) Dual Color, Break Apart Rearrangement Probe. The green and red arrows show split signals, indicating breaks in the ETV6 gene. Yellow arrows show unaltered chromosomes.
 Mucoepidermoid Carcinoma
Mucoepidermoid Carcinoma
Mucoepidermoid carcinoma (ICD–O code 8430/3) is the most common primary malignancy of the salivary gland. A recurring t (11;19)(q21;p13) MECT1-MAML2 translocation is present in a significant number of cases of mucoepidermoid carcinoma.39 This alteration results in a novel fusion oncogene in which the cyclic adenosine monophosphate (cAMP) responsive element–binding (CREB) domain of the CREB–regulated transcription coactivator MECT1 (also known as CRTC1, TORC1, or WAMTP1) is fused to the transactivation domain of the Notch coactivator MAML2 (Fig. 26.2). This fusion protein influences the expression of the cAMP/CREB (FLT1) and Notch (HES1 and HES5) target genes and is then involved in the transformation of epithelial cells.40,41
A t (11;19) and/or an MECT1-MAML2 fusion transcript was detected in more than 55% of the tumors.40 In fusion-negative mucoepidermoid carcinomas, the most common aberrations were single or multiple trisomies.
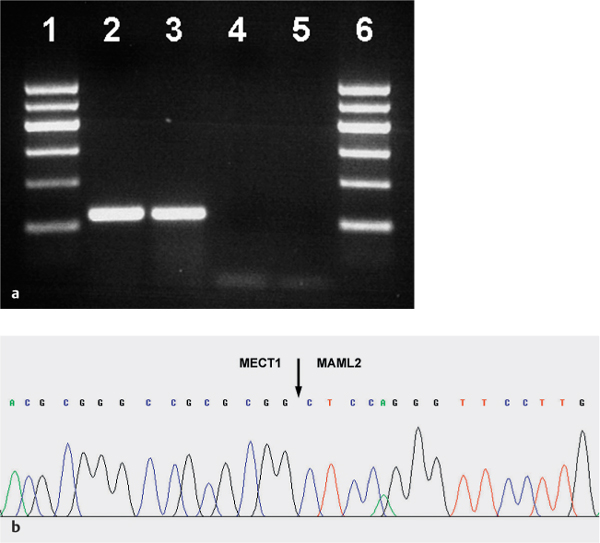
Fig. 26.2 a, b Mucoepidermoid carcinoma
a Expression of the MECT1-MAML2 fusion transcript in a low-grade mucoepidermoid carcinoma, using reverse transcriptase polymerase chain reaction (RT–PCR). Lanes 1, 6: marker (100 bp); lane 2: sample; lane 3: positive amplification control; lane 4: negative amplification control; lane 5: water.
b Sequence analysis of MECT1-MAML2 gene fusion. The translocation breakpoint is indicated by an arrow.
Clinical follow-up studies revealed that fusion-positive patients had a significantly lower risk of local recurrence, metastases, and tumor-related death in comparison with fusion-negative patients. Taking only tumor-related deaths into account, the estimated median survival for fusion-positive patients was over 10 years, compared with 1.6 years for fusion-negative patients. These findings suggest that classifying mucoepidermoid carcinomas molecularly on the basis of MECT1-MAML2 fusion is histopathologically and clinically relevant and that the fusion is a useful marker in predicting the biological behavior of mucoepidermoid carcinomas.40
A novel fusion partner of MAML2 from the CRTC family, namely CRTC3, localized on chromosome 15q26.1, has been found to be present in a subset of mucoepidermoid carcinomas.42 In a recent study, it was shown that CRTC3-MAML2 fusion may be associated with favorable clinicopathological features and that the patients may be younger than those with MECT1-MAML2 fusion and those with no detectable gene fusion.43
Several studies have also detected epigenetic inactivation of the promoter region of various tumor suppressor genes (TSGs).31,32 Recently, Kishi et al.31 reported that promoter methylation of RB1 TSG was associated with shorter overall survival.
 Adenoid Cystic Carcinomas
Adenoid Cystic Carcinomas
A variable chromosomal aberration has been found in adenoid cystic carcinomas (ICD–O code 8200/3). The characteristic molecular feature of this lesion appears to be a recurrent chromosomal translocation, t (6;9)(q22–23;p23–24), which generates a fusion transcript involving the genes for transcription factors MYB and NFIB. Transcription factor Myb consists of an N–terminal DNA–binding domain, a centrally located transcription activation domain, and a C–terminal negative regulatory domain. Myb is highly expressed in immature cells and plays a key role in proliferation, apoptosis, and differentiation. In the t (6;9)(q22–q23;p23–p24) translocation, exon 14 of MYB is fused to the last coding exons of NFIB. This translocation results in deletion of exon 15 of MYB and its 3? untranslated region, which contains several highly conserved target sites for miR-15a/16 and miR-150 microRNAs, which in the normal situation regulate its transcription. It therefore appears that disruption of these regulatory areas leads to overexpression of the fusion transcript.44
Apart from this recurrent translocation, several other rearrangements, including 6q, 9p, 12q, and several other chromosome areas, have been found in individual cases.45–49 Array-based LOH and cytogenetic studies of other chromosomal aberrations in adenoid cystic carcinomas show discordant results. Losses of parts of chromosomes 1p, 6q, 8q, 9p, 12q, and 17p and gains in chromosomes 7p, 17q, and many others have been found (34 and citation therein). Among these, the most frequent losses are located on chromosomes 1p32–36, 6q22–25, and 12q12–13, suggesting putative TSGs in the pathogenesis of adenoid cystic carcinomas in these regions.50–53 The only more remarkable associations between chromosomal changes and clinicopathological features were deletion of 1p32–p36 and loss of heterozygosity at chromosome 6q23–25, which have been associated with a poorer clinical outcome.29,53
It has also been suggested that there is a potential role of inactivation of the tumor suppressor genes CDKN2A and TP53 in the development of adenoid cystic carcinomas and in tumor recurrence and high-grade transformation, respectively.30,54 Frequent methylation of TSG promoter regions has been found in adenoid cystic carcinomas, and these epigenetic alterations are often associated with a higher tumor grade and poorer overall survival.31,55
Several studies have analyzed gene expression profiles. Altered expression of genes related to morphogenesis, neurogenesis, proliferation, apoptosis, extracellular matrix proteins, and basement membrane has been demonstrated. The alterations include overexpression of transcription factors SOX4 and TFAP2C (AP2-gamma), and some genes in the Wnt/β–catenin signaling pathways are probably the most important markers characterizing adenoid cystic carcinoma.24,27
It has also been shown that c-Kit protein overexpression is common finding in adenoid cystic carcinoma.56 Overexpression of c-Kit protein (CD117) may be accompanied by KIT mutation, as has been described for gastrointestinal stromal tumor (GIST) and several other tumor types.57 In adenoid cystic carcinoma, however, the presence of a KIT mutation is controversial. While several studies have shown that KIT mutation is very rare in adenoid cystic carcinoma,56,58 one recent study found that it was present in 88% of the lesions.59 However, the study used formalin-fixed, paraffin-embedded tissues with a very low rate of extraction of amplifiable DNA (57%), so that potential technical artifacts have to be taken into account; further research would be needed to confirm these results.
As tumors with KIT mutation in the regulatory region can be targeted by small-molecule inhibitors of tyrosin kinase such as imatinib mesylate (Gleevec), it has been suggested that they might be used in the treatment of adenoid cystic carcinomas. Patients with adenoid cystic carcinoma showed a remarkable response in only one study, by Faivre et al.,60 but no benefit from imatinib therapy was seen in patients in any of the other studies.61–63 These results appear to correlate with those reported by authors who did not find any mutations in the KIT gene in salivary gland carcinomas. The value of imatinib treatment for patients with adenoid cystic carcinoma is therefore as yet unclear, and further research is needed.
 Polymorphous Low-Grade Adenocarcinoma
Polymorphous Low-Grade Adenocarcinoma
Polymorphous low-grade adenocarcinoma (ICD–O code 8525/3) is a malignant epithelial tumor that occurs only in the minor salivary glands and is characterized by a bland, uniform nuclear morphology and infiltrative growth pattern, with prominent perineural invasion. Polymorphous low-grade adenocarcinoma is clinically characterized by an excellent overall survival rate. Very few cases have been studied using molecular-genetic methods. Early cytogenetic studies showed various chromosomal aberrations.64–66 Surprisingly, cytogenetic similarities between adenoid cystic carcinoma and polymorphous low-grade adenocarcinoma were demonstrated, with frequent chromosome 12 abnormalities and the presence of t (6;12)(p21;q13) translocation in both tumor types.48 Cribriform adenocarcinoma of the tongue has so far been described in only one series67 and has not been studied with genetic methods. It has been postulated that cribriform adenocarcinoma of the tongue is a genuine entity distinct from polymorphous low-grade adenocarcinoma. Cytogenetic and molecular analysis is needed to further characterize the differences between cribriform adenocarcinoma of the tongue and polymorphous low-grade adenocarcinoma.
 Epithelial–Myoepithelial Carcinoma
Epithelial–Myoepithelial Carcinoma
Few cases of epithelial–myoepithelial carcinoma (ICD–O code 8562/3) have been studied. Half of the patients had nondistinctive chromosomal alterations, while the others had normal karyotypes.68
 Clear Cell Carcinoma
Clear Cell Carcinoma
Clear cell carcinoma (ICD–O code 8310/3) is a malignant epithelial salivary gland tumor characterized by monomorphous population of the cells, with optically clear cytoplasm. Very few published data are available on clear cell adenocarcinomas, as there has been controversy regarding the classification of salivary gland tumors with clear cell components. There is a spectrum of distinct entities, such as clear cell myoepithelial carcinoma, hyalinizing clear cell carcinoma, and clear cell adenocarcinoma not otherwise specified. Almost no molecular and cytogenetic studies are available.
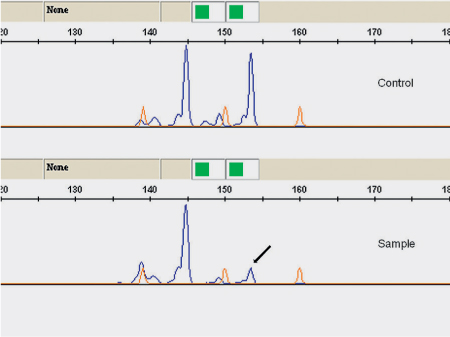
Fig. 26.3 Loss of heterozygosity of marker D16S541 in a case of basal cell carcinoma. The arrow points to the STR allele, with a reduced peak height (ratio < 0.5).
 Basal Cell Adenocarcinoma
Basal Cell Adenocarcinoma
Basal cell adenocarcinoma (ICD–O code 8147/3) is a malignant salivary gland neoplasm composed of basaloid epithelial cells characterized by an invasive growth pattern. Rare molecular cytogenetic studies have shown gains at 9p21.1-pter, 18q21.1-q22.3, and 22q11.23-q13.1, as well as losses at 2q24.2 and 4q25-q27.28 Molecular-genetic studies have found frequent alterations in 16q12–13 regions, with common loss of heterozygosity, in both sporadic and familial basaloid tumors. The study indicates that these tumors share the same alterations as dermal cylindromas and implicates the CYLD gene in their development (Fig. 26.3).69
 Malignant Sebaceous Tumors
Malignant Sebaceous Tumors
No molecular-genetic or cytogenetic data are available for malignant sebaceous tumors (ICD–O code 8410/3).
 Cystadenocarcinoma
Cystadenocarcinoma
No molecular-genetic or cytogenetic data are available for cystadenocarcinoma (ICD–O code 8440/3).
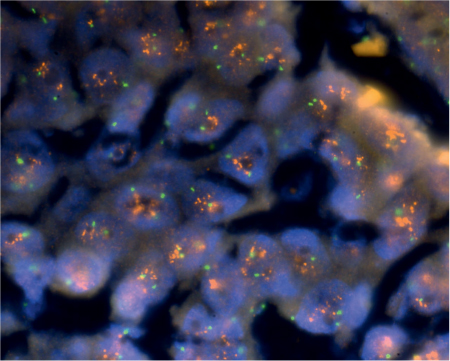
Fig. 26.4 ERBB2 (HER–2/neu) amplification and overexpression in a salivary duct carcinoma. Red signal matches ERBB2 and green signal represents the centromere in the chromosome 17 probe.
 Salivary Duct Carcinoma
Salivary Duct Carcinoma
Salivary duct carcinoma (ICD–O code 8500/3) is an aggressive salivary gland neoplasm that resembles high-grade ductal carcinoma of the breast. In one study, most salivary duct carcinomas had LOH at chromosome 9p21. This chromosomal locus contains the CDKN2A tumor suppressor gene, which has been implicated in the development or progression of salivary duct carcinoma.70
Mutations and overexpression of the TP53 gene and protein, as well amplification of the ERBB2 gene (formerly HER–2/neu) and gene product overexpression are common findings in salivary duct carcinoma (Fig. 26.4). These features are associated with recurrent local disease, distant disease metastasis, and survival time.71,72
 Myoepithelial Carcinoma
Myoepithelial Carcinoma
For myoepithelial carcinoma (ICD–O code 8982/3), comparative genomic hybridization, LOH, and cytogenetic studies carried out in a few patients have shown conflicting results, with variable chromosomal losses.73–76
 Carcinoma Ex Pleomorphic Adenoma
Carcinoma Ex Pleomorphic Adenoma
Carcinoma ex pleomorphic adenoma (ICD–O code 8941/3) develops within a preexisting benign pleomorphic adenoma. As pleomorphic adenoma is characterized by recurrent rearrangement involving transcription factors PLAG1 on chromosome 8q12 and HMGA2 on chromosome 12q15, it is not surprising that these aberrations are also present in carcinoma ex pleomorphic adenoma77,78 (details of these rearrangements are given in Chapter 19).
Gene amplification, manifested as double minute chromosomes and homogeneously staining regions, is present in a substantial proportion of cases of carcinoma ex pleomorphic adenoma. This amplification particularly affects the region on chromosome 12q14–15 in which in particular the MDM2 gene (the regulator of tumor suppressor p53) , HMGA2, and CDK4 are localized.77,79,80 Interestingly, it seems that this genetic event also probably contributes to progression of other types of cancer, as it has also been detected in atypical lipomatous tumors/well-differentiated liposarcoma, for example.81 Amplification of other loci such as ERBB2 has also been identified in carcinoma ex pleomorphic adenoma.80
El-Naggar et al.82 have shown that LOH on chromosome 17p may contribute to progression of carcinoma ex pleomorphic adenoma. Similarly, an LOH study by Fowler et al.83 found alteration of 17p13 in these tumors. These results led to mutational analysis of TP53, which is situated in this region, but with divergent results. While Nordkvist et al.84 found mutations of TP53, other investigators did not identify any association of alterations of p53 with malignant transformation in pleomorphic adenoma.77,85
Several other chromosomal changes have been detected, such as amplifications/gains of 1q11-q32.1, 2p16.1-p12, 8q12.1, 8q22–24.1, and 20, and losses of 1p21.3-p21.1, 5q23.2-q31.2, 8 p, 10q21.3, and 15q11.2. However, few of these appear to be relevant for disease progression.79,80
In general, it appears that amplification of chromosome 12q14–15, where MDM2 and HMGA2 in particular are located, deletions of 5q23.2-q31.2, gain of 8q12.1, where the PLAG1 gene is situated, gain of 8q22.1-q24.1 with the MYC gene, amplification of ERBB2 (HER2/neu), and mutation/overexpression of TP53 may be of importance in the process of malignant transformation that takes place in benign pleomorphic adenoma.
 Although many genetic factors are now known to be associated with the etiology of salivary gland cancer, detection of genetic alterations is not yet being used in clinical routine work for therapeutic and risk stratification of patients with salivary gland cancer.
Although many genetic factors are now known to be associated with the etiology of salivary gland cancer, detection of genetic alterations is not yet being used in clinical routine work for therapeutic and risk stratification of patients with salivary gland cancer.
< div class='tao-gold-member'>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


