Other Environmental Risk Factors
Introduction
Salivary gland tumors are relatively uncommon and there is considerable difficulty in decisions regarding prognosis and management, as well as diagnostic uncertainty that has implications for treatment. The diversity of the lesions is surpassed only by their unpredictable and varied clinical behavior, which poses a unique challenge for clinicians. The etiology of salivary gland tumors is largely unexplained, but there is a possible role for several environmental and genetic factors, and a few risk factors have been identified.
Radiation
There is substantial evidence that exposure of salivary gland tissue to radiation, including therapeutic radiation, is a risk factor (further details are given in Chapter 26). Radiation exposure appears to be important in the development of Warthin tumor, as seen from the increased incidence among atomic bomb survivors.1 Exposure to gamma radiation and x-rays is a known risk factor for both benign and malignant salivary gland tumors. However, the precise mechanism for this adverse effect has not been determined.2
 Radiation exposure is a risk factor for the development of Warthin tumor. The underlying mechanism is unknown.
Radiation exposure is a risk factor for the development of Warthin tumor. The underlying mechanism is unknown.
Viruses
The existence of a tropism for salivary gland tissue, including latent infection of the salivary glands in healthy individuals, is known for several viruses, including Epstein–Barr virus (EBV), cytomegalovirus (CMV), and human herpesvirus (HHV). No specific pathogenic mechanisms or implication of viruses in the development and progression of salivary gland tumors have yet been established. It was recently demonstrated that pleomorphic adenomas contain simian virus 40 (SV40) DNA sequences and express the SV40 large T antigen, suggesting that oncogenic SV40 virus may be involved in the genesis or progression of pleomorphic adenoma.3 Warthin tumor has also been associated with the presence of EBV in the cytoplasm of bilateral tumors.4 Human herpesvirus 8 (HHV8) is the cause of several neoplastic disorders in immunocompromised individuals, especially Kaposi sarcoma. HHV8 is naturally present in human saliva. A high prevalence of HHV8 in patients with Warthin tumors of the salivary glands has been reported, suggesting that the virus has a role in the etiopathogenesis of the tumor.5 Many recent studies have reported that oropharyngeal infection with human papillomavirus (HPV) may be a predisposing factor in the tumorigenesis of squamous cell carcinoma, but little is known about the possible role of HPV in salivary gland tumorigenesis. Recent studies have identified the high-risk HPV16 serotype in oncocytomas, pleomorphic adenomas, and Warthin tumors in the parotid gland in a few cases.6 This initial study of high-risk HPV in a small number of salivary gland lesions has only suggested the possibility of involvement of the virus in the disease, and larger series are needed.
 The precise role of tropism for salivary gland tissue in herpes-viruses, human papillomaviruses, and Epstein–Barr virus in relation to the tumorigenesis of benign tumors is still unclear.
The precise role of tropism for salivary gland tissue in herpes-viruses, human papillomaviruses, and Epstein–Barr virus in relation to the tumorigenesis of benign tumors is still unclear.
Other Environmental Risk Factors
There is considerable evidence for a pathogenetic association between smoking as a risk factor and Warthin tumor, and several recent studies have demonstrated a strong association of smoking with Warthin tumor.7–9 Smokers are eight times more likely to develop Warthin tumor than nonsmokers. The rate of smokers among Warthin tumor patients is up to 90% in men and 88% in women, figures that are much higher in comparison with the rates expected for the normal population of 40% and 32%, respectively.8 A strong correlation between the amount of nicotine intake and the risk for bilateral Warthin tumor has also been documented.8 A strong male predominance for Warthin tumor (more than 90%) was reported in the 1950s.10 With the later increase in the rate of nicotine intake among women, an increased incidence of Warthin tumor was also observed, resulting in a male–female ratio of 1.5: 1.11
There has been persistent controversy over the precise pathogenesis of Warthin tumor. In animal models, chronic nicotine abuse was shown to result in direct morphological and functional changes in salivary gland tissue.12 The morphological and functional disturbances observed are due to chronic nicotine-mediated pathological stimulation of salivary gland tissue. A possible explanation for the nicotine-related tissue damage was suggested in 2000.13 It was postulated that a cigarette smoke–related increase in free radicals ultimately results in oxidative damage to mitochondrial DNA (mtDNA) and a decrease in oxidative phosphorylation.13 In agreement with this hypothesis, a study of alveolar macrophage mtDNA showed that smokers had nearly six times more damage to mtDNA than nonsmokers.14
 Smoking appears to be a significant risk factor for the development of unilateral and bilateral Warthin tumor of the parotid gland.
Smoking appears to be a significant risk factor for the development of unilateral and bilateral Warthin tumor of the parotid gland.
Genetics
Salivary gland tumors are frequently characterized by recurrent chromosome translocations, which have recently been shown to result in pathogenetically relevant fusion oncogenes. These genes encode novel fusion proteins, as well as ectopically expressed normal or truncated proteins, and are found in both benign and malignant salivary gland tumors. The major targets for the translocations in benign salivary gland tumors are DNA–binding transcription factors (PLAG1) and chromatin-associated protein (HMGA2), both involved in growth factor signaling and cell cycle regulation, and coactivators of the Notch (MAML2) and cyclic adenosine monophosphate (MECT1) signaling pathways. Identification of these fusion oncogenes has contributed to our knowledge of molecular pathways leading to epithelial tumors in general, and to salivary gland tumors in particular.15
 Pleomorphic Adenoma
Pleomorphic Adenoma
Pleomorphic adenoma (ICD–O code 8940/0) is the most common benign tumor of the salivary glands. It has marked histological diversity, with epithelial, myoepithelial, and mesenchymal-type cells arranged in a variety of architectural and differentiation patterns.
Extensive cytogenetic studies of pleomorphic adenomas have shown that about 70% are karyotypically abnormal. There are four major cytogenetic subgroups of pleomorphic adenoma: the most common alterations involve 8q12 (40%), followed by tumors with rearrangements of 12q13–15 (8%). Still other pleomorphic adenomas may have sporadic clonal changes at other sites than 12q13–15 or 8q12, and about one-third of cases have a normal karyotype (16 and citation therein).
The target gene on chromosome 8q12 is PLAG1 (pleomorphic adenoma gene 1), which encodes a zinc finger transcription factor. In the vast majority of cases, translocation t (3;8)(p21;q12) with β–catenin as a partner has been found (Fig. 19.1). The other partner genes include LIFR (leukemia inhibitory factor receptor) in t (5;8) (p13q12) translocation and TCEA1 (transcription elongation factor A, SII, 1) in cryptic translocation. The result of these aberrations is promoter swapping, causing deregulation of expression of the PLAG1 gene. Several other cryptic intrachromosomal rearrangements have also been found, giving rise to the novel fusion proteins CHCHD7-PLAG1 and TCEA1-PLAG1.17–20
Activation of the PLAG1 gene on chromosome 8q12 is the most frequent mutation found in pleomorphic adenomas. Abnormal PLAG1 expression results in deregulation of PLAG1 target genes, causing salivary gland tumorigenesis. Plag1 binds to promoter 3 of the insulinlike growth factor 2 gene (IGF2) and stimulates its activity. IGF2 is highly expressed in salivary gland adenomas overexpressing PLAG1, while no IGF2 expression is found in adenomas without abnormal PLAG1 expression or in normal salivary gland tissue, indicating a perfect correlation between PLAG1 and IGF2 expression. These results provide us with the first clue for understanding the role of PLAG1 in salivary gland tumor development.21
In pleomorphic adenomas, both cell populations—epithelial and modified myoepithelial cells—carry PLAG1 rearrangements. This finding further supports the pluripotent single-cell theory, which postulates that the tumor-initiated modified myoepithelial cell evolves into the varied somatic cell phenotypes present in pleomorphic adenoma, and reinforces the role of PLAG1 in the tumorigenesis of benign and malignant pleomorphic adenomas.22
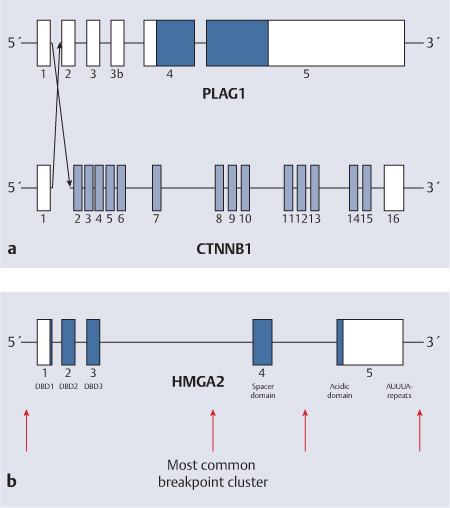
Fig. 19.1 a, b Benign salivary gland tumors targets for translocations.
a Schematic illustration of promoter substitution between the PLAG1 and CTNNB1 genes in pleomorphic adenomas with translocation t (3;8)(p21;q12). The filled boxes indicate coding sequence. The arrows indicate breakpoints.
b Schematic illustration of the HMGA2 gene, breakpoints inside which are indicated by arrows. The filled boxes indicate coding sequence.
The HMGA2 (high mobility group AT–hook 2) gene targeted in a group of pleomorphic adenomas with 12q13–15 rearrangement falls into the family of high mobility group proteins. These are small, acidic, nonhistone, chromatin-associated proteins. HMGA2 contains three DNA–binding domains called AT–hooks, which bind to the minor groove of adenine–thymine-rich sequences, facilitating formation of transcriptional complexes, and an acidic C terminal domain. As the partner gene of HMGA2, the FHIT (fragile histidine triad) gene in translocation t (3;12)(p14.2;q15), the NFIB (nuclear factor I/B) gene in rearrangement ins (9;12)(p23;q12;15), and several other genes involved in complex rearrangement have so far been found in pleomorphic adenomas. The gene fusion results in separation of DNA–binding domains from the mRNA–regulating sequences of HMGA2, leading to deregulation of expression.23–25 Transcription factors Hmga2 and NF–kappa B influence expression of IGF2 through regulation of IGF2BP2 (insulinlike growth factor 2 mRNA binding protein 2, Imp2), and they therefore probably play a role in the development of salivary gland tumors.26–28
 Mutations and deregulations of the PLAG gene family are the most frequent genetic alterations that occur in pleomorphic adenoma.
Mutations and deregulations of the PLAG gene family are the most frequent genetic alterations that occur in pleomorphic adenoma.
< div class='tao-gold-member'>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


