
9

Benign Tumors, Cysts, and Tumor-like Conditions of the Salivary Glands
Three percent of head and neck tumors and 0.5% of all tumors in the body are parotid tumors. Parotid tumors constitute 80% of tumors of the salivary glands, and 80% of parotid tumors are benign.1,2 Five to 10% of salivary gland tumors are in the submandibular gland, and 50 to 60% of these are benign.1,2 Ten to 15% of salivary gland tumors occur in the minor salivary glands, and 20 to 25% are benign.2
 Benign Mixed Tumor (Pleomorphic Adenoma)
Benign Mixed Tumor (Pleomorphic Adenoma)
The use of imaging, fine-needle aspiration, and nerve integrity monitors will be covered in this section on the benign mixed tumor.
Historical Review
The development of parotid surgery can be traced to treatment of the benign mixed tumor. These tumors were first termed mixed tumor by Broca in 1866, and this name was later popularized by Minsenn in 1874.3 Mixed tumor refers to the mixture of epithelial and mesenchymal elements. The term pleomorphic adenoma emphasizes the varied histological presentation of these tumors. Early parotid surgery was performed by Siebold (1793), Billroth (1859), and Virchow (1863).4 Eighteenth-and 19th-century parotid surgery avoided the facial nerve, with intracapsular dissection of the tumor later replaced by extracapsular enucleation. Enucleation was thought to avoid damage to the facial nerve, but it resulted in tumor spillage with associated recurrence rates as high as 45%.5
In 1907 Thomas Carwardine reported facial nerve preservation during parotid surgery.6 Sistrunk7 identified a facial nerve peripheral branch and traced it in a retrograde fashion to the main facial nerve trunk during parotidectomy. Retrograde dissection of the facial nerve was practiced prior to antegrade dissection. Multicentric recurrences were interpreted as being due to a semi-malignant nature of the tumor. McFarland8 drew attention to high rates of recurrence with enucleation and the benign histopathology of these tumors, stating:”The complaisance of the surgeon is not infrequently disturbed by the return of his patient with a recurrence of the tumor, and that of the pathologist, by the continued good health of some patient condemned to death upon the histopathological evidence of a supposed malignancy. “ Patey and Thackray9 advocated wide resection of the tumor by superficial parotidectomy, showing what they termed”focal infiltration of the capsule, and demonstrated how pseudopodia of the tumor could be left behind if the lesion was enucleated.
Not long afterwards it became widely recognized that only the rare tumor with infiltrative and destructive growth had malignant characteristics, and that the vast majority of mixed tumors were in fact benign. The theory that one third of mixed tumors were multicentric was discarded on the discovery that most cases of “multicentricity” were in fact due to tangential cutting of tumor projections and were thus multicentric recurrences of benign mixed tumors. Pathologic review repeatedly revealed pseudopodia perforating the pseudocapsule and projecting into salivary gland tissue. Interestingly, surgeons started performing a wider resection of benign mixed tumor, including facial nerve dissection, because of fears of multicentricity rather than for the more valid reason that enucleation leads to subtotal removal of tumor. More complete parotid surgery led to higher rates of temporary and permanent injury to the facial nerve, postoperative hematoma, gustatory sweating, and facial contour depression. To avoid these complications and reduce recurrences, a return to enucleation followed by radiation therapy was practiced in some centers.10 The risk of radiation-induced malignancy led to the abandonment of this modality.
Surgery for mixed tumor of the parotid has evolved from enucleation to retrograde and subsequent ante-grade facial nerve dissection with adequate margin of normal parotid parenchyma except where the tumor abuts the facial nerve. Modern skillfully trained surgeons perform parotidectomy with a low risk of permanent facial nerve dysfunction and recurrence.
Presentation
The benign mixed tumor is the most common salivary gland neoplasm in adults and children, with most occurring in the parotid gland.1,11 Eighty-five percent of mixed tumors present in the parotid gland (Fig. 9–1). Two thirds of parotid neoplasms are mixed tumors.2 They present in all ages, with the highest incidence in the fourth decade of life.1 They present more commonly in females.1 They are firm except in a tumor that is predominantly myxoid (hypocellular). Extrinsic pressure can result in facial nerve dysfunction on rare occasion. The biological characteristics do not vary by age, and low recurrence rates are expected after appropriate resection.
Parotid mixed tumor is most often diagnosed and treated when the tumor is small (<4 cm), mobile, and located in the superficial lobe. Eighty percent of the parotid parenchyma is lateral to the facial nerve. Ninety percent of parotid mixed tumors present in the superficial lobe, and 80% are located in the lower pole. Ten percent of mixed tumors extend to or are limited to the deep lobe of the parotid. They may also present very rarely in the accessory parotid gland. The accessory parotid gland, present in up to 56% of individuals, connects to the main excretory duct of the parotid gland and is found on the anterior surface of the main parotid gland or anterior to the parotid.12
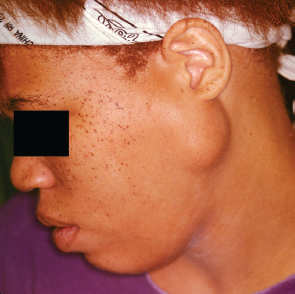
FIGURE 9-1 Parotid mixed tumor
Parotid deep lobe tumors, including mixed tumors, may extend above or below the stylomandibular ligament in the space between the angle of the mandible and styloid process and present as a parapharyngeal space mass. The stylomandibular ligament runs with three muscles inserted in the styloid process: the styloglossus, stylohyoid, and stylopharyngeus. It inserts in the posterior border of the ascending ramus and angle of the mandible. Tumors that extend anterosuperior to the stylomandibular ligament are dumbbell tumors that are constricted between the mandible, stylomandibular ligament, and skull base. Tumors extending posteroinferior to the stylomandibular ligament are rounded in appearance. Parapharyngeal tumors, including mixed tumors, can result in sleep apnea from associated intraoral swelling and medial displacement of the tonsil and lateral pharyngeal wall, often presenting without trismus. The carotid space vessels and cranial nerves (CN IX–XII) of the post-styloid compartment of the parapharyngeal space are not involved with the tumor as deep lobe parotid tumors present in the prestyloid compartment. Paragangliomas and most schwannomas are found in the poststyloid compartment of the parapharyngeal space. Intraoral biopsy of parapharyngeal mixed tumors will result in tumor seeding and scar tissue, making definitive trans-cervical resection more difficult.
Mixed tumors are most often solitary tumors with a rare presentation as a synchronous or metachronous tumor in the same gland or contralateral gland. Uncommonly, they can present together with another salivary gland tumor, most commonly a Warthin’s tumor, but also with a mucoepidermoid, acinic cell or adenoid cystic carcinoma.13
Five percent of mixed tumors occur in the submandibular gland.2 Fifty to 60% of submandibular tumors are mixed tumors, presenting as a painless swelling.14 Ten percent of mixed tumors occur in minor salivary glands, and only very rarely do they occur in the sublingual gland.2 More than half of benign tumors of the minor salivary glands are mixed tumors.2 They most commonly occur as painless, submucosal masses in the hard and soft palate, with equal incidence, but without ulceration. In the palate they usually present lateral to the midline. They have poor mobility when on the hard palate because of the underlying bone. They may also present in the upper lip and buccal mucosa, where they are mobile.2 Mixed tumors can arise in the nasal cavity and paranasal sinuses. Paranasal sinus mixed tumors may cause nasal obstruction or obstructive sinusitis. Pharyngolaryngotracheal minor salivary gland neoplasms, including mixed tumors, may present with hoarseness, dysphagia, cough, or hemoptysis.
Esoteric heterotopic salivary gland rests can develop mixed tumors in the cervical lymph nodes, ear, lacrimal gland, mandible, sellar region, lung, breast, trunk, and lower limbs.15 In the skin, mixed tumors are called chondroid syringomas. Chondroid syringomas present from the fourth through sixth decades more commonly in males. The tumors have been found in most areas of the skin, but the majority present in the skin of the face and head. They can originate from sebaceous glands, sweat glands, or ectopic salivary glands.16
Imaging
Ultrasound can differentiate a benign mixed tumor from a malignant tumor in over 90% of cases.17 Criteria for malignancy include irregular shape, ill defined and irregular margins, inhomogeneous structure, and the presence of abnormal lymph nodes. High-resolution probes and harmonic imaging can demonstrate histopathological heterogeneity of mixed tumors in many cases17(see Chapter 2 for a full discussion).
Computed tomography (CT) and magnetic resonance imaging (MRI) may predict mixed tumor if lobular shape and homogeneous internal echoes are present. CT imaging is less expensive but more prone to degradation artifact than MRI. CT should be performed with contrast enhancement. MRI with gadolinium enhancement is superior at demonstrating the internal structure of the salivary gland and delineating tumor from normal salivary gland. MRI has different signal intensity for tumor, fat, and muscle. An MRI of a mixed tumor typically shows postcontrast enhancement, a high T2 signal, and well-defined margins (unless large enough to be lobulated) that do not invade surrounding tissue planes.18 CT or MRI will demonstrate deep lobe parotid tumor extension to the prestyloid compartment of the parapharyngeal space. The prestyloid compartment is anterior to the carotid space vessels. The deep lobe parotid tumor displaces the parapharyngeal fat medially. Deep lobe parotid tumors extending into the parapharyngeal space can be seen connected to the parotid and can be distinguished from minor salivary parapharyngeal tumors that are completely surrounded by fat (see Chapter 2, Figs. 2–13, 2–14).
Positron emission tomography with fluorine-18 fluorodeoxyglucose has not yet proven useful in classification of salivary gland neoplasms as benign or malignant. A false-positive rate of 31% has been reported in one study.19
Pathogenesis
The pathogenesis of the benign mixed tumor is uncertain. The multicellular theory hypothesizes that salivary gland tumors develop from the proximal and distal ectodermally derived differentiated cell types in the adult salivary gland (acinar-duct subunit). The mixed tumor in this theory comes from the distal intercalated and myoepithelial cells and consists of epithelial and myoepithelial elements. The myoepithelial cells are responsible for the extracellular matrix.20,21
The bicellular or reserve theory suggests that basal cells of excretory ducts and intercalated ducts are stem cells from which adult salivary gland units and tumors derive.22 The mixed tumor would derive from intercalated duct stem cells in this theory.23
Cytology
Fine-needle aspiration of benign mixed tumor usually yields benign epithelial cells admixed with stroma (Fig. 9–2). Varying degrees of cellularity and differing proportions of epithelium and stroma are seen. The epithelial cells may be arranged as sheets, cords, or single cells. The stromal component is acellular, fibrillary material that is metachromatic (magenta-purple) with Romanovsky-type stains (e. g., Diff-Quik or Giemsa, which are commonly used for rapid evaluation of specimen adequacy).24 A typical benign mixed tumor is usually not a diagnostic dilemma. Occasional cases may be highly cellular with minimal stromal component, or contain cystic changes, squamous metaplasia, or necrosis that can mimic other neoplasms. Squamous metaplasia in benign mixed tumor can be mistaken for squamous cell carcinoma.
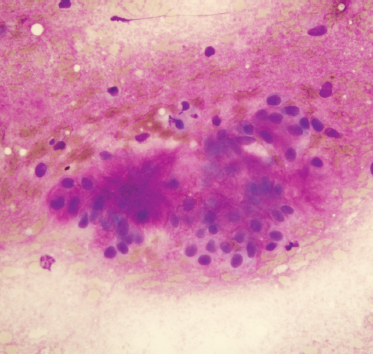
FIGURE 9-2 Fine-needle aspiration of benign mixed tumor, containing benign oval epithelial cells and pink-purple fibrillary matrix material (Diff-Quik stain, ×400).
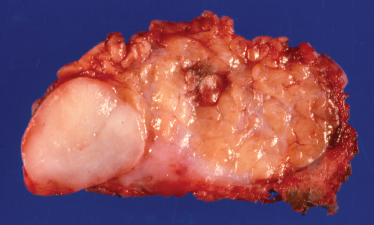
FIGURE 9-3 Gross photograph of benign mixed tumor showing circumscription of the mass.
Alterations in histology following fine-needle aspiration can result in exuberant squamous metaplasia, infarction and necrosis, subepithelial stromal hyalinization, acute and chronic hemorrhage with inflammation with multinucleate giant cells, granulation tissue with subsequent fibrosis, cholesterol cleft formation, pseudoxanthomatous reaction, pseudocapsular invasion, and microcystic degeneration.
Histology
Mixed tumors of the parotid are benign epithelial tumors with an incomplete fibrous capsule of varying thickness. Mixed tumors in the minor salivary glands usually do not have a capsule.24 Even nonencapsulated mixed tumors show distinct circumscription of the lesion on gross examination and low-power microscopy (Fig. 9–3). Protuberances give a lobulated appearance as the tumor grows. Mixed tumors are round, smooth, freely movable, and grossly gray to yellow in color.
Microscopically, mixed tumors are biphasic neoplasms that contain benign epithelial and myoepithelial cells with a variety of patterns, and mesenchymal stroma (Fig. 9–4). The term pleomorphic adenoma was coined to reflect the large variety of patterns these tumors can assume. The epithelial cell forms are predominantly salivary duct and myoepithelial cells. Closely associated nonductal cells include spindle, round, stellate, plasmacytoid, polygonal, and clear forms. Rarer cell forms include keratinized squamous epithelium, oncocytes, basal cells, and sebaceous cells or goblet cells. The stromal component is usually myxomatous or myxochondromatous, but hyaline, fibrotic, or osseous (boneforming) areas may also be present in the stroma.24 It is believed that the stroma is produced by the myoepithelial cells.25 Tumors may be epithelial-cell rich (cellular) (Fig. 9–5) or stromal rich (myxoid) (Fig. 9–6). Tumors are more highly cellular (the epithelial component predominates) in their early stages of development, and the amount of chondromyxoid stroma (the mesenchymal component) increases with the duration of the neoplasm.23 Recurrent mixed tumors are frequently more hypocellular (stromal-rich) with associated higher rates of incomplete encapsulation.26,27
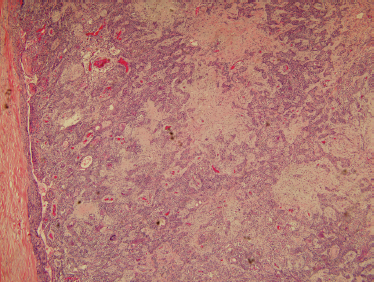
FIGURE 9-4 Histology of usual benign mixed tumor showing a biphasic neoplasm with epithelial and stromal components (H&E, ×40).
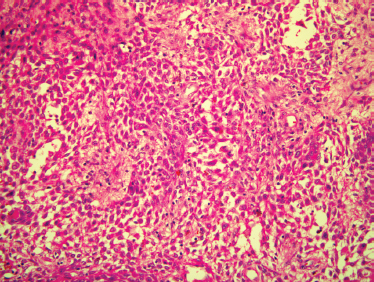
FIGURE 9-5 Histology of cellular benign mixed tumor. These tumors predominantly contain the epithelial component of a benign mixed tumor, with only scant amounts of chondromyxoid matrix (H&E, ×200).
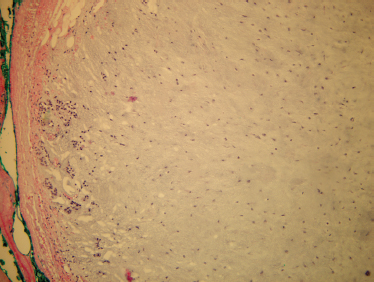
FIGURE 9-6 Histology of stroma-rich benign mixed tumor. These tumors are predominantly myxoid stroma, with only a sparse epithelial component (H&E,×100).
Genetics
The first observation of recurrent loss of a chromosome14 was in a meningioma. The mixed tumor was the second type of benign tumor for which nonrandom chromosomal changes were reported.28 The mixed tumor has been cytogenetically well characterized. In addition to the cytogenetic subgroup with an apparently normal diploid stemline (making up 30% of the cases), three major cytogenetic subgroups with abnormal karyotypes can be distinguished. By far the largest cytogenetic subtype consists of tumors with chromosome 8 abnormalities mainly showing translocations involving band q12 (8q12). Patients with the 8q12 abnormality tend to be younger, and they have more hypercellular tumors. The ectopic expression of the PLAG1 gene is associated with benign mixed tumor in individuals with 8q12 translocation. The other subgroups involve various translocations involving 12q15 and a group of nonrecurrent clonal abnormalities.28 Another reported chromosome abnormality involves chromosome bands 3p21.29 There is thus far no correlation between these cytogenetic characteristics and recurrence.
The mutation of the tumor suppressor gene p53 is the most common genetic alteration in human cancers. Immunohistochemistry of the p53 and the proliferative marker Ki-67 has been absent from primary parotid and submandibular mixed tumors indicating that mixed tumors of the parotid and submandibular gland are histologically similar and that they have a low proliferative rate and good prognosis.14
The degree of deoxyribonucleic acid (DNA) instability as revealed by the immunohistochemical staining with anti-single-stranded DNA antibody after acid hydrolysis (DNA instability test) can be used as a marker of malignancy. Twenty-one of 33 (64%) mixed tumors were positively stained by DNA instability test diffusely or sporadically, indicating that mixed tumors can be regarded as “unstable” and often contain or predispose to malignant subclones with occasional capsular or extracapsular invasion, reflecting the potential progression to malignancy. In comparison, no sign of potential malignant progression was identified in the seven Warthin’ tumors studied.30
Treatment
The informed consent for the removal of a parotid mixed tumor should include transient or permanent facial nerve dysfunction, numbness, gustatory sweating, seroma, hematoma, and recurrence. The parotid mixed tumor is most commonly treated with partial superficial parotidectomy (dissection of the facial nerve with a 2 cm margin of normal parotid parenchyma except where the tumor abuts the facial nerve),31 or complete superficial parotidectomy with facial nerve dissection. The facial nerve is most commonly dissected in an antegrade fashion, but a retrograde approach is still preferred by some and is not associated with a higher rate of complications, including facial nerve dysfunction.32 The more variable course of the peripheral facial nerve branches has mostly resulted in abandonment of this technique. Total parotidectomy33 and extracapsular dissection34 have also been used for benign mixed tumor. Extracapsular dissection differs markedly from other parotid procedures because facial nerve dissection is not performed. Extracapsular dissection is contrasted to enucleation by its advocates as dissection of a small cuff of normal parotid parenchyma just outside the capsule of the parotid tumor.34
Regardless of the extent of normal parotid parenchyma that is resected with the tumor, there will be a near universal focal capsule exposure where the facial nerve or superficial fascia abuts the tumor31,35. A positive margin with partial superficial parotidectomy or complete superficial parotidectomy will occur in up to one third of cases because of pseudopodia piercing the pseudocapsule where the tumor abuts the facial nerve.31,35 Few separations of pseudopodia from the main tumor occur with expertly performed contemporary parotid surgery because most of the tumor has a margin of normal parotid parenchyma. Enucleation leads to more positive margins with unacceptable recurrence rates. Capsular penetration occurs where the tumor forms protrusions into the pseudocapsule, sheared off with enucleation.
Partial superficial parotidectomy (Fig. 9–7) for small (>4 cm), mobile tumors of the lateral lobe will result in less transient facial nerve dysfunction, gustatory sweating (Frey’s syndrome), facial depression, and operative time compared with procedures (total parotidectomy and complete superficial parotidectomy) that resect a greater portion of normal parotid parenchyma and dissect more of the facial nerve.31 Extracapsular dissection if improperly performed is enucleation and will result in unacceptable recurrence rates. If a normal cuff of parotid parenchyma is resected without dissection of the facial nerve, a potentially higher risk of permanent facial nerve dysfunction may ensue. A meta-analysis summary effect for permanent facial nerve dysfunction is reported 1.8 times higher for extracapsular dissection compared with superficial parotidectomy.31 Additionally, the exact procedure indicated for a parotid mass cannot always be determined in the preoperative setting, and successful surgery may require wide exposure with facial nerve dissection, not afforded in extracapsular dissection.
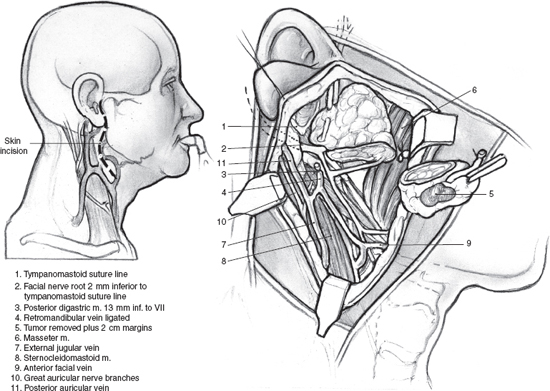
FIGURE 9-7 Partial superficia parotidectomy. Note the mean distance from the tympanomastoid suture to the facial nerve is 2 mm. The mean distance from the posterior belly of the digastric muscle to the facial nerve is 13 mm.
Mixed tumors extending to the parapharyngeal space (Fig. 9–8 See also Chapter 2, Fig. 2–13) can usually be removed with a transcervical approach after superficial parotidectomy and facial nerve dissection (see Chapter 15). Selected larger, retromandibular mixed tumors approaching the skull base may require mandibulotomy. Lip split, closure of intraoral structures, and midline extension of the cervical incision can be avoided with a vertical mandibulotomy posterior to the lingula of the medial surface of the mandible, preserving the inferior alveolar nerve and intraoral sensation and providing a panoramic view of the parapharyngeal tumor (Figs. 9–9, 9–10). Modern plating systems should be contoured and applied prior to the mandibulotomy. They are then removed and reapplied after mandibulotomy and resection of tumor. This approach will minimize the risk of nonunion, malunion, or malocclusion.
The treatment of a submandibular mixed tumor is resection of the gland along with the tumor with a margin of normal tissue where possible. The informed consent should include trauma to the marginal mandibular branch of the facial nerve potentially resulting in a temporary or permanent lower lip asymmetry, intraoral numbness, and trauma to the hypoglossal nerve resulting in tongue deviation, infection, and hemorrhage. Rare recurrences are possible after this approach. The treatment of a minor salivary gland mixed tumor is resection of the gland with a margin of normal tissue where possible. The informed consent would vary with the anatomical presentation.
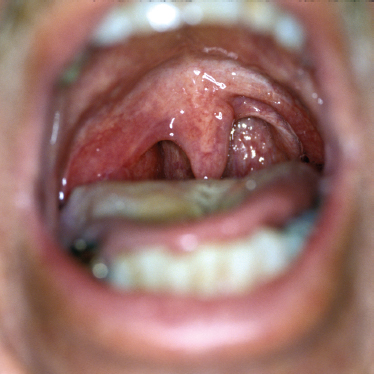
FIGURE 9-8 Right parapharyngeal tumor.
Recurrence
Most cancer tumor registries do not track mixed tumors; therefore, follow-up remains deficient. Recurrence rates for mixed tumors are also difficult to evaluate because of the small number of patients and the variability of follow-up times. In most series, parotidectomy with facial nerve dissection results in recurrence rates of 0 to 4%.31 Recurrences generally occur in the first 10 years, with a mean interval to the first recurrence of 7 years.36 Late recurrences beyond 20 years occur, although rarely. Recurrent mixed tumors are almost always multinodular.37 Imaging studies coupled with clinical exam will document the multiplicity of recurrence over clinical exam alone. Seventy-five percent of recurrences are in the superficial lobe (See Chapter 2, Fig. 2–12).38 Time intervals between operation and recurrence are significantly shorter for enucleation compared with parotidectomy and facial nerve dissection.39 Patients with more than one recurrence tend to have their first recurrence earlier (mean 47 months) than patients who were cured after reexcision of their only recurrence (mean 105 months).40
The chief factor for tumor recurrence is enucleation (Table 9-1), where pseudopodia of tumor extending beyond the pseudocapsule are sheared off.36,39–41 The reported meta-analysis summary effect reveals a 9 times higher rate of recurrence for enucleation compared with superficial parotidectomy.31 Recurrence is also possible, but much less likely in parotidectomy with facial nerve dissection in areas where the tumor abuts the facial nerve and a “partial” enucleation is performed.31
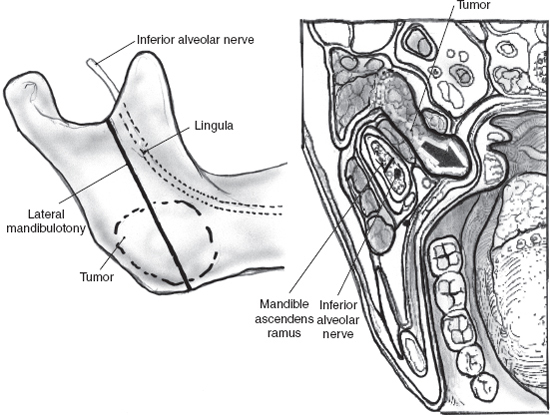
FIGURE 9-9 Vertical mandibulotomy posterior to the lingula of the medial surface of the mandible.
The other major significant reason for recurrence is tumor rupture and spillage. The overall rate of recurrence using superficial parotidectomy with facial nerve dissection is 2.6% in a review of 23 publications with 2366 total patients. When the capsule is ruptured using superficial parotidectomy with facial nerve dissection, the rate of recurrence is 5% (p<. 05).31 The percentage of mixed tumors that recur during enucleation is 30%.31 This high rate is because of the heightened incidence of tumor rupture as well as the shearing of tumor pseudopodia that pierce the pseudocapsule. Older, hypocellular tumors (myxoid-type tumors), with focal absence of encapsulation, are also more friable and prone to rupture26,27 Hypocellular tumors show greater focal absence of encapsulation (compared with cellular tumors), with tumor merging into normal parotid gland tissue in 70% of cases.27
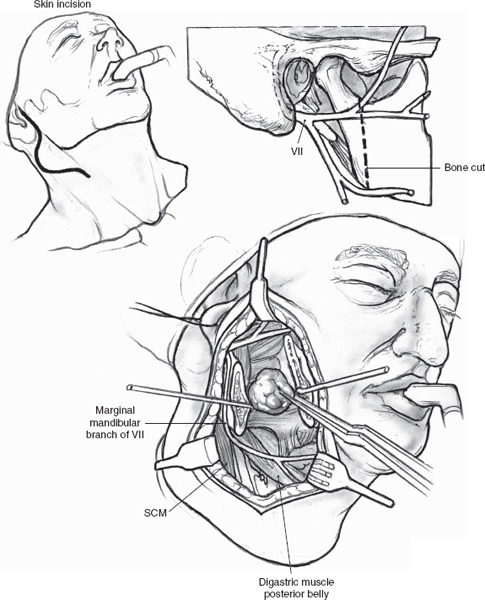
FIGURE 9-10 Panoramic view after vertical mandibulotomy. SCM, sternocleidomastoid muscle.
Table 9-1 Risk Factors for Recurrence of Benign Mixed Tumors
| 1. Enucleation |
| 2. Tumor ruptur |
| 3. Hypocellular tumors |
| 4. Deep lobe/parapharyngeal tumors |
| 5. Large tumors |
| 6. Younger/female patients |
| 7. Previously recurrent tumors |
| 8. Increased proliferative activity |
Other predisposing factors to recurrence include deep lobe tumors that are at an increased risk for rupture.42 Removal via the perioral approach will increase the chance of rupture and recurrence.40 Higher recurrence rates are not uniformly reported in younger and females33,42,43 patients but are a factor in some series.40 Recurrence rates are higher for previously recurrent tumors.44 Subsequent recurrence after an initial recurrence occurs at a rate of 25%.45 If there is one recurrence, additional recurrences can be expected at a shorter interval.41 The true primary multicentric mixed tumor is very rare.39
Tumors in the upper neck attached to the inferior aspect of the parotid gland can be mistakenly diagnosed as lymph nodes and are therefore at times enucleated through an inadequate incision. A watchful waiting approach under this clinical circumstance will inevitably lead to a high rate of recurrence. This situation is managed with superficial parotidectomy with nerve dissection.46
Recurrences may also rarely occur in adequately resected tumor. In these cases there may be a characteristic of the tumor that increases its chance for recurrence. Flow cytometric DNA analysis suggests high S-phase fractions are associated with larger tumor size and tendency to recur.47 Large tumors may recur because of difficulty in their removal, their increased proliferative activity, or for both reasons.
Immunohistochemical staining of pathologic specimens of mixed tumor has also allowed the evaluation of the expression of cell proliferation associated nuclear antigen (Ki-67). The expression of Ki-67 has been correlated with mitotic activity, histological grade, and clinical behavior of tumors, including salivary gland tumors. Proliferation markers are low in hypocellular, myxoid tumors, both primary and recurrent, but the proliferation activity is higher in the uncommon cell-rich recurrent tumors compared with its primary presentation.37,48 Proliferation activity is markedly higher in epithelial cells of recurrent mixed tumors compared with nonrecurrent tumors. Immunhistochemical markers on recurrent tumors are also higher for progesterone receptor compared with primary tumors.49
The differential diagnosis for a recurrent mixed tumor is a malignant transformation of a previously benign mixed tumor, a neuroma, or a lymph node. Fine-needle aspiration can frequently make the diagnosis. A spectrum of treatment options has been promoted for recurrent mixed tumors. Observation can be appropriate in the debilitated or elderly patient, particularly with evidence of benign tumor on fine-needle aspiration. Observation has also been advocated in selected cases if the recurrence is nonprogressive and asymptomatic, waiting for the presentation of the expected multiple sites of recurrence and therefore avoiding multiple surgeries.
Recurrent mixed tumors are multinodular in up to 98% of cases when clinical exam is supplemented with imaging.38 Resection of a localized lesion close to the thin layer of fibroblasts that surround the tumor will likely result in later recurrence.50 Unencapsulated nodules are often embedded in surrounding healthy fat tissue and scar tissue, so that total parotidectomy with facial nerve dissection and resection of scar is the first line of treatment for recurrent mixed tumor.37 Retrograde facial nerve dissection, particularly the likely undisturbed temporal facial nerve branch, has an important potential role in recurrent mixed tumor. Total parotidectomy with facial nerve sacrifice and grafting is an option exercised in selected cases.45 Resection and reconstruction of overlying skin with a pectoralis myocutaneous flap or radial forearm free flap may be required.
Radiotherapy after surgery for an isolated recurrence in a patient younger than 40 years of age is not indicated. Hearing impairment and malignant degeneration after radiation therapy must be considered. However, treatment of the patient with multicentric and multiple recurrences, particularly in an individual older than 40 years of age, can include surgery followed by radiation therapy. Treatment of recurrent multinodular disease with surgery alone compared with surgery and postoperative radiation resulted in a reduction in subsequent recurrence from 43 to 4% in one series.51 Neutron radiotherapy is superior to conventional radiotherapy for malignant salivary gland tumors, and it appears promising in patients with multiply recurrent mixed tumors who are not candidates for surgery.52 Local-regional control remains less both in conventional and neutron radiotherapy for patients with gross residual disease compared with microscopic disease. Radiotherapy will not be effective if a large mixed tumor load remains (see Chapter 13).
Facial nerve injury after surgery for recurrent mixed tumors occurs in up to 40% of cases and the rate increases with each revision procedure.51 The chance of new relapse and facial nerve injury will be higher in patients treated with prior parotidectomy with nerve dissection than enucleation.38 Infiltration of the facial nerve is 8 times greater with recurrent tumors.53 Deep lobe recurrences, recurrences in multiple sites, and extensive scar tissue will increase the rate of facial nerve injury.54 Intraoperative monitoring of the facial nerve for recurrent tumors is appropriate.
The potential devastation of recurrence cannot be underestimated. One third of patients with recurrent tumors do not ultimately achieve tumor-free status.40 Recurrent tumors, although benign, can behave (biologically) as malignant tumors, recurring in the deep parotid lobe, neck, or base of the skull, and present formidable or incurable situations.
 Myoepithelioma
Myoepithelioma
Myoepitheliomas, benign tumors composed almost entirely of myoepithelial cells and a minor stromal element, comprise 1% of salivary gland neoplasms. Most myoepitheliomas occur in the parotid gland (40%), followed by the hard and soft palate (21%), and rarely in the submandibular gland.55 There is no gender predilection. They are painless, slow-growing tumors.
Myoepithelial cells lie beneath the ductal and acinar epithelium, typically lining the basement membranes of the salivary glands. These cells combine features of both smooth muscle and epithelial cells.56 Myoepithelial cells also occur in sweat glands, mammary glands, and the prostate. They contract, helping to express secretions from glandular acini to secretory ducts in the salivary glands.57
In the multicellular theory, tumors develop from ectodermally derived differentiated cell types in the adult salivary gland (acinar-duct subunit), with distal cellular elements consisting of intercalated and myoepithelial cells composed of epithelial and myoepithelial elements. Myoepithelioma may represent one end of the spectrum of mixed tumor when one considers that both myoepithelial cells and ductal/acinar epithelial cells may derive from a single stem cell precuror in salivary glands.56 This theory is substantiated by a shared chromosome 12q cytogenetic abnormality in these two types of neoplasms.57
Myoepitheliomas are smooth-surfaced tumors with a white-colored interior. They have a thin capsule in the parotid gland and are unencapsulated when located in the palate. Microscopically, myoepitheliomas may exhibit solid, myxoid, or reticular growth pattern. The myoepithelial cells may appear spindled, epithelioid, clear, or plasmacytoid58 (Fig. 9–11). The histological variants do not have prognostic significance. Spindle cell variants are the most common.59 Ductal structures are either absent or very rare. Some investigators state that one or more ductal structures at medium (×200) to high (×400) power field excludes a diagnosis of myoepithelioma, and suggest that even rare ductal structures mean that the tumor is a cellular benign mixed tumor.56 Others state that no more than 5 to 10% of cells should have duct structures for a diagnosis of myoepithelioma.60 Most authors feel that the myxochondroid stroma of benign mixed tumor should be absent in myoepithelioma, and, if present, the lesion should best be regarded as a benign mixed tumor.61 As this discussion illustrates, the distinction between cellular benign mixed tumor and myoepithelioma is somewhat arbitrary.
Myoepitheliomas must be distinguished from other tumors with spindle cells, including extracranial meningioma, schwanomma, paraganglioma, fibroma, and leiomyoma. The plasmacytoid variant must be distinguished from a plasmacytoma. Immunohistochemical studies are positive for cytokeratins, vimentin, and S-100 protein and help confirm the myoepithelial nature of the tumor.56 The rare malignant myoepithelioma may arise from a preexisting mixed tumor or a benign myoepithelioma or more commonly de novo. Malignant transformation is often preceded by a longer clinical course of multiple recurrent benign tumors.56 Accumulation of p53 protein, perhaps through mutational events, may play a role in malignant transformation.62
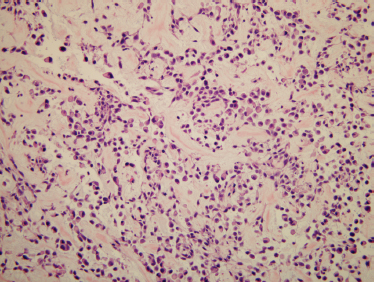
FIGURE 9-11 Histology of myoepithelioma. The cells in this example are plasmacytoid-like, although many other patterns are possible. Immunostaining was done on this neoplasm and showed the cells to be myoepithelial cells (positive staining for cytokeratin and S-100; H&E, ×200).
The treatment of benign myoepithelioma is similar to the mixed tumor, given the similar biologic behavior of the tumor. Recurrence after partial or complete superficial parotidectomy is rare, occurring less commonly than with the mixed tumor.59 Resection of the palatal presentation would include a cuff of normal tissue.
 Warthin’s Tumor
Warthin’s Tumor
The American pathologist Aldred Scott Warthin vividly described two cases as papilliferous in 1929.63 His eponym is widely used, although Hildebrand has the earliest report in the 19th century.64
Presentation
Warthin’s tumor, or papillary cystadenoma lymphomatosum, is generally regarded as a true neoplasm confined almost exclusively to the parotid gland. It is the second most common benign neoplasm of the parotid gland, making up 20 to 30% of parotid tumors and generally occurring between the fourth and seventh decade.65 The tumor presents as a slow-growing mass, often in the tail of the parotid, although it can become inflamed with a painful presentation and rapid growth after an indolent course for years. Warthin’s tumors presenting in minor salivary glands many times are discovered in the lip or palate. They are occasionally found in the parapharyngeal space and submandibular gland. Warthin’ tumor can be rarely found in the larynx, lacrimal glands, and nasopharynx.55
Multifocal Warthin’s tumor occurs in up to 20% of patients.66 The bilateral presentation in 5 to 6% of cases is a more common finding than in any other salivary gland tumor.66 Bilateral occurrence has been noted less frequently for oncocytoma, acinic cell carcinoma, and basal cell adenoma.67 Extraglandular presentation of Warthin’s tumor in the neck makes up 3 to 5% of Warthin’s tumor.68 There are sporadic case reports of Warthin’s tumor associated with mixed tumor, oncocytoma, or lymphoma.
Warthin’s tumor is more common in male patients. An increasing number of female patients are being reported with Warthin’s tumor closely correlated with the increase in smoking rates noted in female patients. Thirty to 40% of patients with Warthin’s tumor are now female. Over 90% of patients with Warthin’s tumor smoke, supporting a correlation between cigarette smoking and Warthin’s tumor65,69 Warthin’s tumor. Warthin’s tumor has been considered rare in African Americans; however, an increasing percentage of African Americans are being reported with this tumor.69 Warthin’s tumor is also rare in Africa.70 Ionizing radiation has been associated with benign and malignant tumors. Study of atomic bomb victims in Hiroshima and Nagasaki, Japan, demonstrates an increased frequency of Warthin’s tumor with increasing radiation dose.71
Imaging
Nuclear imaging is helpful in Warthin’s tumor and oncocytic tumors. Technetium Tc99m pertechnetate uptake is due to the epithelial component (oncocytic portion) of Warthin’s tumors, and tumors with a large epithelial component and lesser amounts of cystic spaces show a larger radioactive index.72 An increased uptake of the isotope is not noted in all Warthin’s tumors, and a negative scan does not exclude this tumor.73 Normal salivary glands and Warthin’s tumor concentrate radioactive iodine. This needs to be considered on evaluation of diffentiated thyroid cancer for metastasis.
The tumor contents of Warthin’s tumor are best imaged with MRI (See Chapter 2, Fig. 2–9). Unilateral, but particularly bilateral, nonenhancing tumor, with well-defined margins on MRI, with a high T2 signal, is likely to be Warthin’s tumor.18 Preoperative imaging with MRI (MRI has superior soft tissue resolution) or CT may determine multifocality or extraparotid origin of Warthin’s tumor.
Warthin’s tumor may present on imaging as a cystic parotid lesion. Using ultrasound, hypoechoic areas may represent the cystic component of Warthin’s tumor. Other cystic parotid lesions include benign lymphoepithelial lesions, branchial cleft cysts, chronic sialadenitis, cystic low-grade mucoepidermoid carcinoma, cystic mixed tumor, lymphangioma, and lymphoma. For unknown reasons positron emission tomography may result in high fluorine-18-fluorodexyglucose (FDG) accumulation in the parotid gland in patients with Warthin’s tumor.74
Pathogenesis
The pathogenesis of Warthin’s tumor is not fully understood (Table 9-2). The parotid gland is the first to develop embryologically and the last to be encapsulated. The most popular hypothesis on the pathogenesis of Warthin’s tumors is the incorporation of salivary ducts in lymphatic tissue during late encapsulation.75 Another hypothesis is that the tumor arises from heterotopic salivary ducts within preexisting lymphoid tissue or periparotid lymphoid tissue.76 In the reserve cell theory, Warthin’s tumor derives from intercalated ducts. It has also been proposed that Warthin’s tumor is not a neoplasm, but rather a metaplastic process with a secondary lymphoid reaction. The nonclonal nature in one study of Warthin’s tumor determined by the polymerase chain reaction method suggests that Warthin’s tumor may be a nonneoplastic tumor-like condition.35 The higher incidence of autoimmune disorders (Hashimoto’s thyroiditis, autoimmune hyper-and hypothyroidism) in patients with Warthin’s tumor and the higher rates of smoking promote an autoimmune or inflammatory role in the formation of Warthin’s tumor.77 Warthin’s tumors that are predominantly epithelial in cellular consistency are smaller than the classic or lymphoid predominant tumors, suggesting that Warthin’s tumor may initially develop from an adenomatous epithelial proliferation followed by lymphocytic infiltration.78
Table 9-2 Theories of the Genesis of Warthin’s Tumor
| 1. Warthin’s tumor results from the incorporation of salivary ducts in lymphatic tissue during late encapsulation |
| 2. Warthin’s tumor arises from heterotopic salivary ducts within preexisting lymphoid tissue or periparotid lymphoid tissue. |
| 3. According to reserve cell theory, Warthin’s tumor derives from intercalated ducts. |
| 4. Warthin’s tumor is not a neoplasm but rather a metaplastic process with a secondary lymphoid reaction. |
| 5. Warthin’s tumor may initially develop from an adenomatous epithelial proliferation followed by lymphocytic infiltration. |
| 6. Epstein-Barr virus genome is an inciting factor. |
| 7. Enzyme type 2 nitric oxide synthase is a stimulating factor. |
Lymph nodes are found within the parotid gland, and these nodes can contain salivary tissue. The late encapsulation of the parotid may be why lymph nodes adjacent to but separate from the parotid may also contain salivary gland tissue, explaining the finding of salivary tumors in the upper neck, not in continuity with the parotid gland. Warthin’s tumor may result from a proliferation of these salivary gland rests.79
Using in situ hybridization techniques, the Epstein-Barr virus genome was first detected in the cytoplasm of neoplastic cells of multiple or bilateral Warthin’s tumor at a rate of 87% compared with 17% for solitary Warthin’s tumor. This suggested a strong association between infection of cells with this virus and the development of multiple or bilateral Warthin’s tumor.80 Other evidence refutes Epstein-Barr virus as a cause of Warthin’s tumorogenesis. Ogata et al81 found Epstein-Barr DNA in 62% of Warthin’s tumors; however, in situ hybridization for Epstein-Barr ribonucleic acid (RNA) showed that the nuclei of the neoplastic epithelial cells of all tumors were negative. Although in situ hybridization for Epstein-Barr virus DNA revealed that the nuclei of the neoplastic epithelial cells were positive in 4 of the 21 tumors, the positive cells were sparsely distributed, and there was no evidence of monoclonal proliferation of Epstein-Barr-positive neoplastic epithelial cells.81
Immunohistochemical studies have identified prolonged nitric oxide production by the enzyme type 2 nitric oxide synthase implicated in the pathogenesis of many solid tumors. Nitric oxide and type 2 nitric oxide synthase are known to be associated with p53. A significant association between enzyme type 2 nitric oxide synthase and p53 staining is identified in Warthin’s tumors.82
Cytology
The typical cytologic features of Warthin’s tumor are oncocytic epithelial cells and lymphocytes (Fig. 9–12). The epithelial cells are usually present in cohesive sheets. The lymphocytes are cytologically benign and may be predominantly small mature lymphocytes or a heterogeneous lymphoid population with mixed small and large lymphocytes, corresponding to follicle formation (germinal centers) in the lymphoid stroma. Many Warthin’s tumors have a cystic component, and aspirated cyst contents may have a brown, turbid,”motor oil” gross appearance. Microscopically, the cyst fluid contains degenerated cells, proteinaceous fluid, and cholesterol clefts.83 Various metasplastic changes may occur in the epithelium of Warthin’s tumor, including squamous metaplasia, mucinous metaplasia, and sebaceous differentiation.
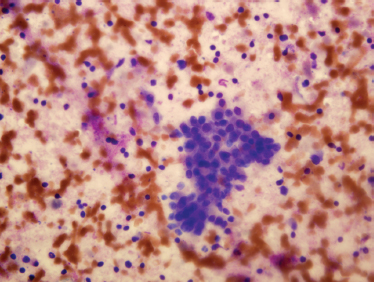
FIGURE 9-12 Fine-needle aspiration of Warthin’s tumor, showing mixed oncocytes (the larger, cohesive cells) and background small benign lymphocytes (Diff-Quik stain,×400).
Fine-needle aspiration of Warthin’s tumor may be nondiagnostic if only the cyst contents are aspirated. It is important to sample a residual solid mass if a cyst is aspirated. CT-guided fine-needle aspiration may reduce sampling error, avoiding the pitfall of a nondiagnostic aspiration of the cystic component of the salivary gland mass. If a lymphocyte-rich area is aspirated, an erroneous diagnosis of a lymphoproliferative lesion such as lymphoma may be entertained. If predominantly oncocytic epithelium is sampled, it may be difficult to distinguish Warthin’s tumor from oncocytoma. Squamous metaplasia in Warthin’s tumor may be mistaken for squamous cell carcinoma or mucoepidermoid carcinoma.84
Postaspiration histological changes of Warthin’s tumor can include infarction, fibrosis, and exuberant squamous metaplasia.
Histology
Warthin’s tumors are encapsulated with a smooth or lobulated surface. Cystic areas on gross cross-sectioning may contain a brownish fluid. Warthin’s tumor consists of oncocytic epithelium, frequently with papillary architecture, with a lymphoid stroma and cystic spaces. The columnar oncocytic epithelium is bilayered with oxyphilic granular cells (Fig. 9–13). The oncocytic cells contain excessive mitochondria that show frequent structural abnormalities and reduced metabolic function. An increase in mitochondrial DNA damage secondary to smoking has been hypothesized.85 Immunohistochemistry using antimitochondria antibody may help in the identification of oncocytic cells.86 Metaplastic (infarcted) Warthin’s tumor is characterized by replacement of the original oncocytic epithelium by squamous metaplasia along with extensive necrosis, fibrosis, and inflammatory change that can be misinterpreted for malignancy. Metaplastic Warthin’s tumor may occur spontaneously or after fine-needle aspiration.87
FIGURE 9-13 Histology of Warthin’s tumor. These lesions show bilayered oncocytic epithelium and a dense, lymphoid stroma (H&E, ×40).
Four subtypes of Warthin’s tumor have been characterized. Subtype 1 has an epithelial tumor component of 50%. It occurs in 77% of cases. Oncocytic differentiation and focal metaplasia to goblet cells or squamous epithelium are found. Subtype 2 is lymphoid stroma poor with an epithelial component of 70 to 80%. It appears like an oncocytoma in areas and occurs in 14% of cases. Subtype 3 is lymphoid stroma rich with an epithelial tumor component of only 20 to 30%. Subtype 3 is found in 2% of cases. In subtype 4, large areas of squamous cell metaplasia and regressive changes are identified. This subtype is seen in 7% of cases. The different subtypes are not associated with different biologic behaviors.88
Immunohistochemistry of cytokeratin expression for Warthin’s tumor and its metaplastic variant indicates that both express cytokeratins 7, 8, 18, and 19, typical for columnar differentiation. The expression of cytokeratins 5/14 and 17, which are typical of regenerative cells, is restricted to basal cells in Warthin’s tumor but is expressed in basal and also in surface cells in metaplastic Warthin’s tumor.89
Genetics
Warthin’s tumor has been successfully karyotyped, and clonal numerical and/or structural changes have been detected in some of these tumors. Two recurrent abnormalities include the 6p rearrangements and t(11;19) in almost half of Warthin’s tumor cases.90 Clonal alterations in this study support that Warthin’s tumor is a true neoplasm rather than an autoimmune-or hypersensitivity-related tumor-like condition.90
The follicular lymphoid infiltrate in Warthin’s tumor is polyclonal when examined by polymerase chain reaction technology. T-and B-cell markers indicate normal lymphoid populations in Warthin’s tumor. DNA is largely diploid.
Treatment
A highly accurate diagnosis of Warthin’s tumor by fine-needle aspiration may prompt conservative management in selected cases. An overall diagnostic accuracy rate of 70% by fine-needle aspiration and not higher suggests that histological confirmation is still necessary in many cases.91 Enucleation of Warthin’s tumor has been advocated to reduce morbidity,92 but without imaging studies it increases the risk of failing to remove multicentric tumor. Partial superficial parotidectomy and complete superficial parotidectomy are standard treatment options.93 Wide exposure and careful palpation are recommended to appreciate multicentricity. Total parotidectomy has been promoted because of multicentricity. Many clinicians do not advocate long-term follow-up,93 but metachronous Warthin’s tumors even after prolonged follow-up has led some to advocate long-term-follow up.79 Extraglandular Warthin’s tumors make up 3 to 5% of Warthin’s tumors and are treated surgically with few recurrences.79 Malignant transformation of Warthin’s tumor most often to mucoepidermoid carcinoma is reported in 1% of cases.94
 Basal Cell Adenoma
Basal Cell Adenoma
In older classification schemes, all benign salivary gland neoplasms that lacked chondomyxoid stroma were termed monomorphic adenomas. The various types of tumors that fell under this heading were split out as different named types of monomorphic adenomas in the 1970s, and by the time of publication of the second Armed Forces Institute of Pathology fascicle, eight different types of monomorphic adenomas were recognized.83 Basal cell adenoma was one of these tumor types. When the World Health Organization revised the classification system once again in 1991, the umbrella term monomorphic adenoma was dropped, many of the subtypes were renamed, and several were grouped under the term basal cell adenoma.
Basal cell adenomas make up 2 to 5% of salivary gland tumors. Seventy-five percent occur in the parotid and 5% in the submandibular gland. The upper lip is the most common presentation of a minor salivary gland basal cell adenoma, followed by the buccal mucosa.55 There is a female predominance of this tumor by a 2:1 ratio. Basal cell adenoma occurs later in life, when compared with a mixed tumor, with a peak incidence in the seventh decade of life. Such tumors rarely occur in children. They appear as solitary masses, excluding the membranous subtype, which is often multifocal.95 They are hard to palpation, apart from the basal cell adenoma with a large cystic component. They are mobile, except when located on the hard palate.
Basal cell adenomas are solid, well-circumscribed tumors that have a pinkish brown or gray appearance on cut surface. They are encapsulated in the parotid but unencapsulated in minor salivary glands. On aspiration, they are composed of small, basaloid cells with a micro-acinar architecture. Basal cell adenoma is distinguished from mixed tumor by the absence of the chondroid and myxoid foci that typify mixed tumors and facilitate its recognition on fine-needle aspiration55 (Fig. 9–14). Fine-needle aspiration may distinguish basal cell adenoma from mixed tumor, cyst, lipoma, or lymph node. The most difficult cytological distinction is between basal cell adenoma and other tumors with a predominance of basaloid cells, such as cellular mixed tumor, basal cell adenocarcinoma, and adenoid cystic carcinoma. The distinction between basal cell adenoma and cellular mixed tumor is of little clinical importance, but distinction from the malignant tumors is a serious clinical issue. Clinical factors of facial nerve dysfunction or imaging studies showing invasion point to an adenoid cystic carcinoma. The location in a minor salivary gland would also favor adenoid cystic carcinoma, given that minor salivary glands often host adenoid cystic carcinoma.
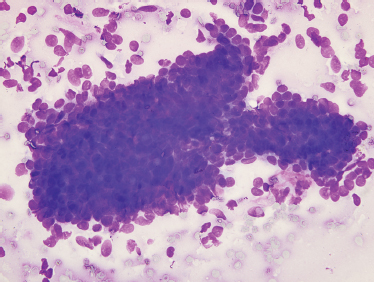
FIGURE 9-14 Fine-needle aspiration of basal cell adenoma. Aspirates of these lesions may show a variety of patterns. All contain small, basophilic cells without fibrillary matrix material(Diff-Quik stain, ×400).
Cytological features of the cell stroma interface are useful in distinguishing basal cell adenomas of the solid type and the adenoid cystic carcinoma. The collagenous stroma in basal cell adenomas interdigitates with adjacent cells in basal cell adenoma. In adenoid cystic carcinoma the two are separated by a sharp, smooth border. Given the difficulty distinguishing a basal cell adenoma from the solid form of adenoid cystic carcinoma by fine-needle aspiration, along with the divergent clinical implications, the diagnosis is best left to the histology.96 On histology the distinction is more evident, with basal cell adenomas being circumscribed and not invading surrounding tissue.
Histology shows basaloid cells in a variety of architectural arrangements with a minimal amount of collaganized stroma, but without the abundant myxoid matrix that is required for the diagnosis of benign mixed tumor (Fig. 9–15). Subtypes of basal cell ade noma include tubular, trabecular, solid, and membranous or dermal analogue tumor.55 Membranous basal cell adenomas are typically located in the parotid gland and are known for a high recurrence rate in part because they are not encapsulated. Bilateral occurrence is reported for the membranous subtype but is not seen in the tubular or other subtypes of basal cell adenoma. The membranous basal cell adenomas have a high rate of malignant transformation, giving rise to basaloid adenocarcinoma ex-monomorphic adenoma.97 They not infrequently occur in association with dermal adnexal lesions. Membranous basal cell adenoma has a clinical and histological resemblance to dermal cylindroma, sharing similar incidence of alterations at the 16q12 13 region, supporting a common molecular origin.98
FIGURE 9-15 Histology of basal cell adenoma. The tumor is composed of small nests of basaloid cells surrounded by basement membrane material (H&, ×200).
Treatment of basal cell adenoma is excision with a normal cuff of surrounding tissue. They uncommonly recur (with the exception of the membranous subtype) when treated by superficial or partial superficial parotidectomy with nerve dissection. The multifocal nature of membranous basal cell adenoma suggests treatment with total parotidectomy.97 Submandibular or minor salivary gland tumors should be treated with resection of the submandibular gland with the tumor or resection of the minor salivary gland tumor with a rim of normal tissue.
 Canalicular Adenoma
Canalicular Adenoma
Canalicular adenomas were separated from monomorphic adenomas and basal cell adenomas in 1991, and they are now classified separately from them.99 They arise almost exclusively from minor salivary glands and seldom occur outside the oral cavity. Like basal cell adenomas, canalicular adenomas are found more commonly in females and in an older population of patients in their seventh decade of life. These slow-growing asymptomatic masses occur most commonly in the upper lip and are the second most common upper lip salivary gland tumor after the mixed tumor.55 The second most common site is the buccal mucosa, and they rarely are found in the parotid. They can be multifocal.100 They are encapsulated with branching and interconnecting cords of double cell thick rows of bland, basaloid, cuboidal to columnar epithelium in a loose stroma without mesenchymal tissue.15 Myoepithelial cells are usually not part of the tumor, and thus immunostains for smooth muscle actin or smooth muscle myosin are negative.101 This can be helpful in the distinction between canalicular adenoma and other benign adenomas, such as benign mixed tumor or basal cell adenoma. Treatment is superficial or partial superficial parotidectomy. Recurrences are rare.
 Oncocytoma
Oncocytoma
Oncocytomas usually present in the sixth through ninth decade of life as a solitary parotid mass with rare multicentric and bilateral occurrence. They have an equal gender distribution. They account for 1% of salivary gland neoplasms, originate from oncocytes, and are found mostly in the parotid gland, occasionally in the submandibular gland, and rarely in the minor salivary glands. They can be bilateral in up to 7% of cases.102 When associated with radiation exposure, they occur 20 years earlier than the mean age for patients with oncocytoma not exposed to radiation.103
Oncocytes are epithelial cells with accumulations of mitochondria. They are found predominantly in salivary gland tissue but also in the thyroid, parathyroid, respiratory tract, pituitary, pancreas, and the kidney. Oncocytic cells in the salivary glands can be categorized as oncocytic metaplasia, oncocytosis, and oncocytoma. Oncocytic metaplasia is a transformation of acinar and ductal cells to oncocytes. This is a phenomenon associated with aging and most often occurs after age 50. Oncocytic metaplasia occurs in oncocytomas, mixed tumors, and mucoepidermoid carcinoma (Table 9-3). Oncocytosis is the proliferation of oncocytes in the salivary glands either diffusely or in foci that produce microscopic or macroscopic nodules. An oncocytoma is a clinical entity, larger than focal oncocytosis with at least partial encapsulation.15 In the multicellular theory, oncocytic tumors derive from striated duct cells.
Table 9-3 Where Oncocytic Cells, Epithelial Cells with Accumulations of Mitochondria, Are Found
| 1. Oncocytoma |
| 2. Clear cell oncocytoma |
| 3. Mixed tumor |
| 4. Warthin’s tumor |
| 5. Malignant oncocytoma |
| 6. Adenoid cystic carcinoma |
| 7. Mucoepidermoid carcinoma |
| 8. Adenocarcinoma |
| 9. Distant metastasis from thyroid carcinoma or renal cell carcinoma |
| 10. Oncocytic metaplasia |
| 11. Oncocytosis |
On fine-needle aspiration, a monotonous population of enlarged cells with abundant, granular cytoplasm is seen (Fig. 9–16). In surgical specimens, most oncocytoma are well circumscribed and encapsulated. Minor salivary gland oncocytomas, in contrast, are not encapsulated and have less defined borders. Oncocytomas are composed of sheets of oncocytes that are granular eosinophilic cells as a result of abundant mitochondria (Fig. 9–17). A true oncocyte has no lymphoid cells, differing therefore from the Warthin’s tumor.55 Other tumors with oncocytes must be excluded, including the benign mixed tumor, malignant oncocytoma, adenoid cystic carcinoma, mucoepidermoid carcinoma, and adenocarcinoma.15 Benign oncocytoma has a lower Ki-67, a measure of proliferative activity determined by immunohistochemistry, than the Ki-67 of a malignant oncocytoma.104 Metastatic renal cell tumor and thyroid tumors that contain a large number of oncocytes also must be differentiated from primary salivary gland oncocytoma.15
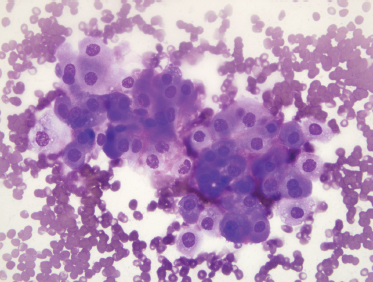
FIGURE 9-16 Fine-needle aspiration of oncocytoma. The smear shows a monotonous population of oncocytic cells with abundant granular cytoplasm (Diff-Quik, ×400).
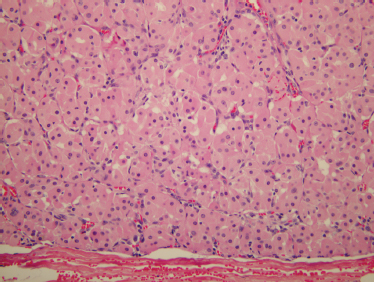
FIGURE 9-17 Histology of oncocytoma. These encapsulated tumors are usually solid and contain large cells with pink granular cytoplasm (H&E, ×200).
Radionucleatide scanning with technetium 99m pertechnetate results in uptake and retention. Most parotid and submandibular cases of oncocytoma are predictable and readily cured by surgery. Multifocal growth pattern and incomplete excision account for recurrent cases.
A variant of oncocytoma is clear cell oncocytoma, in which glycogen is also present in the cells, and the mitochondria are marginated to the edges.105 The presence of glycogen imparts a clear appearance to the cells of hematoxylin-eosin (H&E) staining. The differential diagnosis of clear cell oncocytoma is different from traditional oncocytoma and includes other clear cell tumors, such as metastatic renal cell carcinoma, sebaceous neoplasms, and mucoepidermoid carcinoma.106 The biologic behavior of clear cell oncocytoma is the same as for traditional oncocytoma.
Treatment of parotid and submandibular oncocytomas is complete surgical resection, and most follow a benign outcome. Oncocytic tumors of the minor salivary glands are distinctive from the major salivary gland tumors. Minor salivary gland oncocytomas are less predictable, grow in irregular patterns, and can be locally invasive to cartilage and bone. Most of these rare tumors arise from the minor salivary glands of the false vocal folds and ventricles. They may occur at any site in the laryngeal mucosa. They present as a painless mass in patients over 50 years of age, often in individuals with a smoking history. There is no gender predilection. Hoarseness, cough, and airway obstruction are possible when located in the larynx. On physical exam they appear as a polypoid laryngeal mass. The tumor can be multifocal with local destruction of laryngeal cartilage even when the neoplasm does not have any malignant features. CT imaging can be similar to a cyst of the laryngeal saccule. Grossly, they present as smooth and round cysts. They can have numerous small cysts with papillary projections and can have a similar histological appearance to a Warthin’s tumor minus the lymphoid stroma. Classic Warthin’s tumor can also present rarely in the larynx. They are part of a spectrum of clinically benign cystic and papillary lesions. They are derived from oncocytic metaplasia and hyperplasia of minor salivary gland ducts, referred to as papillary oncocytic cystadenoma.15 There is debate as to whether these oncocytic lesions represent true neoplasms. Treatment is endoscopic surgical excision. They generally do not recur after excision. Because multifocal presentation is possible, close follow-up is recommended. Oral cavity oncocytomas can be oncocytic papillary cystadenomas107 or more solid oncocytomas.108 Sinonasal oncocytomas are locally aggressive and characterized by multiple recurrences.109
 Cystadenoma
Cystadenoma
These unusual multicystic (and occasionally unicystic) neoplasms with papillary proliferations are more common in females, with presentation most commonly in the fifth decade of life. Half of these tumors occur in the minor salivary glands, mostly of the lips and buccal mucosa. Parotid masses make up most of the remaining neoplasms, with 5% occurring in the submandibular glands.55 They are slow-growing asymptomatic masses. They are often encapsulated and are multicystic masses with papillary fronds covered by bland epithelium.24 Treatment is excision with adequate margin.
 Ductal Papilloma
Ductal Papilloma
These rare benign adenomas of ductal epithelium feature a papillary growth. The three ductal adenoma subtypes in order of frequency are: (1) intraductal papilloma,(2) sialadenoma papilliferum, and (3) inverted ductal papilloma. They derive from the excretory duct.
Intraductal papilloma is an asymptomatic papillary proliferation causing dilatation of the duct. These asymptomatic, submucosal neoplasms occur in the fifth and sixth decade of life. They are unicystic (most cystadenomas are multicystic). They distend the duct wall with a papillary proliferation of duct epithelium. They mostly form near the mucosal surface of the excretory ducts of the oral minor salivary glands, with the lip and palate being the most common locations. They are often encapsulated. Their treatment is excision with rare recurrence.110
Sialadenoma papilliferum is a very rare tumor that can be clinically mistaken for a squamous papilloma. It occurs in adults, with a slight male predominance. Most of these neoplasms occur in the hard and soft palate, often at the junction, followed by the buccal mucosa.111 These well-circumscribed, painless, submucosal tumors occur near or at the orifice of the excretory duct near the oral mucosa. The biphasic growth pattern consists of an outer portion with exophytic finger-like papillary projection and an inner endophytic component of glands and ducts.112 The sialadenoma papilliferum is morphologically similar to the papillary syringoadenoma of the sweat gland.113 It is treated with surgery. Recurrence rates of 10 to 15% assert a more significant biological behavior than inverted ductal papilloma and intraductal papilloma.112
Inverted ductal papilloma also is a luminal papillary proliferation that arises at the junction of the duct and oral mucosa. These minor salivary gland neoplasms present predominantly in the lip and buccal mucosa and have no gender predilection. They occur in adults in their fourth to sixth decade of life. They appear to arise from the excretory duct at a deeper level than the intraductal papilloma.112 They are circumscribed, painless, submucosal masses that grow in an inverting pattern forming a broad-based mass. Histologically, they resemble the inverted papilloma of the nose and paranasal sinuses. They are treated with surgical resection and rarely recur. They are not associated with malignant degeneration.
Ductal papillomas must be distinguished from other neoplasms with papillary growth pattern, including Warthin’s tumor, cystadenoma, mucoepidermoid carcinoma, acinic cell carcinoma, and cystadenocarcinomas.
 Sebaceous Lymphadenoma and Sebaceous Adenoma
Sebaceous Lymphadenoma and Sebaceous Adenoma
Intraoral sebaceous differentiation, mostly in the buccal mucosa and vermilion border of the upper lip, is expected in 80% of the population, where these glands are known as Fordyce’s granules. Approximately 10% of parotid and submandibular glands have sebaceous differentiation.114 Sebaceous glands are also occasionally found in periparotid lymph nodes. There are no morphologic differences between cutaneous sebaceous glands and the sebaceous glands in a salivary gland neoplasm. These sebaceous cell collections can be found in association with sebaceous neoplasms of the salivary glands.
The sebaceous lymphadenoma is a rare tumor presenting as a painless, slow-growing tumor mostly in the parotid, but occasionally in the minor salivary glands, and even more rarely in the submandibular gland. They occur mostly in patients in the fifth through seventh decade of life. They derive from sebaceous glands located at the blind ends of intralobular ducts. They can be encapsulated both in the parotid and minor salivary glands. Histological exam reveals islands of round-shaped, small, well-differentiated squamous epithelial cells, with focal sebaceous differentiation, lining the walls of cysts.115 A background of lymphoid stroma with germinal centers is present, suggesting that these neoplasms arise from sebaceous glandular rests in a lymph node similar to what one observes in Warthin’s tumor. Surgical excision is the treatment, and recurrence is not expected.
Sebaceous adenomas are very rare tumors presenting as a painless mass, most commonly in the fifth and sixth decade of life. There is a slight male predominance. About 50% occur in the parotid, with most of the rest presenting in the minor salivary glands and a few in the submandibular gland. They are encapsulated neoplasms composed of sebaceous cell rests with squamous differentiation, without lymphoid follicles. Surgical excision is the treatment, and recurrence is not expected.55
 Mesenchymal Parapharyngeal Tumors
Mesenchymal Parapharyngeal Tumors
Tumors of the parapharyngeal space include deep lobe parotid tumors, neurogenic tumors, paragangliomas, and lymphoma.
Neurogenic Tumors: Schwannomas
The schwannoma is the most common parapharyngeal space neurogenic tumor, constituting 20 to 30% of tumors of the parapharyngeal space.116 Schwannomas or neurilemmomas arise from the Schwann’s cell of the nerve sheath. Only the optic and olfactory nerves lack Schwann’s cells. Twenty-five to 45% of all schwannomas occur in the head and neck.117 After the vestibular schwannoma (acoustic neuroma), the parapharyngeal space is the most common site of a nerve sheath tumor.117 In the parapharyngeal space they may arise from CN IX, X, XI, and XII, the third division (mandibular) of CN V, the sympathetic nerve trunk, and the upper cervical nerves.118 Most parapharyngeal schwannomas present in the poststyloid compartment arising either from the sympathetic chain or vagus nerve, but also CN IX, XI, and XII.116 Schwannomas arising from the lingual, inferior alveolar, and auriculotemporal nerve may occur in the prestyloid space.
Schwannomas are slow-growing, painless, well-encapsulated, solitary tumors associated with the peripheral nerve of origin. In comparison to intratemporal schwannomas with bony confines and limited space available for tumor extension and early neurological sequelae from nerve compression, parapharyngeal schwannomas can reach a large size before causing symptoms. The female to male gender ratio on presentation is about 2:1. They present commonly between 30 and 60 years of age. They can displace the pharynx and tonsil medially on intraoral exam or present as a cervical mass.118 Because of stretching and compression of surrounding structures, the associated neurological deficit may not correlate with the nerve of origin.
Imaging is used to detect schwannomas, vascular tumors (paragangliomas), and parotid tumors and to detect if the carotid artery is at risk. CT or MRI studies can usually distinguish between a schwannoma and a paraganglioma118 (see Chapter 2, Fig. 2–14). Both most often present in the poststyloid space. Schwannomas exhibit strong enhancement on gadolinium-enhanced T1-weighted imaging. On T2-weighted imaging with high spatial resolution, facial nerve schwannomas appear as hypointense, round masses.119 Schwannomas enhance less than paragangliomas and lack the serpiginous flow voids (“salt and pepper”) enhancement pattern of a paraganglioma.118 Most prestyloid para-pharyngeal masses are deep lobe parotid tumors. MRI, CT, and arteriography cannot distinguish between a prestyloid schwannoma (arising from the lingual, inferior alveolar, or auriculotemporal nerve) and a deep lobe parotid tumor.118
Fine-needle aspiration may be performed for para-pharyngeal tumors, usually to distinguish between a parotid epithelial neoplasm such as benign mixed tumor versus a neurogenic tumor. Although benign mixed tumor is usually a straightforward diagnosis, it is difficult to diagnose neurogenic tumors by fine-needle aspiration and to distinguish between benign and malignant neural tumors. In addition, fine-needle aspiration may cause significant bleeding if the tumor is a paraganglioma. For this reason, imaging prior to surgical resection without fine-needle aspiration is a recommended approach for a poststyloid parapharyngeal mass.
If fine-needle aspiration of schwannoma is performed, aspirates may yield spindle-shaped cells with wavy nuclei. Occasionally, nuclear palisading is seen.120 Histologically, schwannomas contain Antoni type A tissue with a palisading array of nuclei around a central mass of cytoplasm (Verocay bodies) and×or Antoni type B with a loose surrounding stroma with no distinctive fiber and cell pattern (Fig. 9–18). Malignant degeneration is rare.
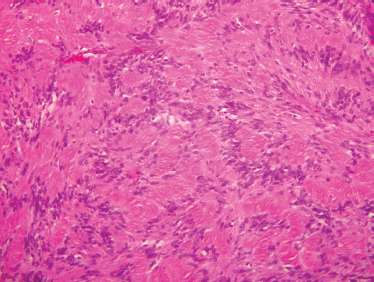
FIGURE 9-18 Histology of schwannoma. These benign neural tumors often show palisaded nuclei (Antoni A areas). (H&, ×200).
Transcervical surgical approach with or without mandibulotomy (see Chapter 15) is preferred over a transoral approach.118 A subplatysmal skin flap with preservation of the marginal mandibularis branch of the facial nerve is followed by ligation of the facial artery and vein. The submandibular gland is retracted anteriorly, and the posterior belly of the digastric is skeletonized or divided. The stylomandibular ligament is released, and access to the parapharyngeal space is achieved. Parotidectomy and removal of the styloid process can enhance exposure for superior extension.
Schwannomas arise eccentrically from the outer surface of the nerve sheath and can be dissected from the nerve of origin. This is an oversimplification, as the tumor can be intimately involved with the nerve. Dissection of most sympathetic chain schwannomas results in substantial injury to the nerve.118 This clinically manifests as Horner’s syndrome. Vagal schwannomas can extend into the jugular foramen and into the posterior cranial fossa, resulting in multiple cranial nerve dysfunctions. Arteriography and subsequent infratemporal, transmandibular, and transpterygoid approaches may be required.
Schwannomas of the peripheral facial nerve are rare, with most presenting in the intratemporal portion of the facial nerve. They can reach a large size prior to becoming symptomatic. Gradual onset facial paralysis occurs in 20% of cases.121 Diagnosis may be suggested by strong enhancement on MRI on gadolinium-enhanced T1-weighted imaging and fine-needle aspiration. Nonetheless, preoperative diagnosis is not always established. Surgery of extratemporal facial neuromas will require a meticulous dissection of the facial nerve, frequently resulting in nerve damage. This intraoperatively results in a dilemma for the surgeon treating the patient who has normal preoperative facial nerve function. In the patient with an established preoperative diagnosis of peripheral facial nerve schwannoma, the patient must be advised of the likely outcome of facial nerve grafting. Postponing surgery may delay nerve injury, but as the tumor enlarges, eventual excision will increase the likelihood of facial nerve dysfunction. Radiation therapy may have a potential role.
Paragangliomas
Paragangliomas or glomus tumors derive from islands of neural crest cells comprising part of the diffuse neuroendocrine system or amine precursor and uptake decarboxylase (APUD) system. The diffuse neuroendocrine system includes a group of cells throughout the body that produce and secrete neurotransmitters converting biologic amines such as dopa and dopamine to neurotransmitters, including norepinephrine and epinephrine. The conversion of norepinephrine to epinephrine is catalyzed by phenylethanolamine-N-methyltransferase that exists only in the adrenal medulla and in a few neurons in the central nervous system. Head and neck paragangliomas, therefore, do not secrete epinephrine, but do accumulate norepinephrine. All paragangliomas have neurosecretory granules seen by electron microscopy, but only 1 to 3% are functional.122
Approximately 90% of tumors arising from paraganglia are pheochromocytoma from the adrenal gland, where most chromaffin cells are located. Most extra-adrenal tumors are located in the abdomen, with a smaller number from the thorax. Three percent of extra-adrenal tumors are located in the head and neck. Eighty percent of head and neck paragangliomas concentrate in the carotid bifurcation (carotid body tumors).123 In descending frequency, the adventitia of the jugular bulb (superior ganglion glomus jugulare) and ganglion nodosum (inferior ganglion glomus vagale) of the vagus nerve, followed by the middle ear (glomus tympanicum), are the other locations of head and neck paraganglioma. They present usually as solitary tumors. Ten percent of cases are multiple, presenting with paragangliomas of the neck, or neck combined with adrenal or extra-adrenal site.124 They can be associated with multiple endocrine neoplasia type II (pheochromocytoma, medullary thyroid carcinoma, and hyperparathyroidism) and neurofibromatosisI. 125 type Ten percent of patients have a family history of paraganglioma, and in these patients one in four will have multiple paragangliomas.126 Chronic hypoxic stimulation from living at high altitude is a contributing cause.
Paragangliomas often present in the fourth through sixth decades of life. They may be asymptomatic or present with hoarseness, dysphagia, aspiration from vocal cord paralysis, tongue hemiatrophy, palatal weakness, Horner’s syndrome, and pain. The superior jugular ganglion paraganglioma (glomus jugulare) can extend beneath the skull base into the parapharyngeal space, having both an intracranial and extracranial component. The extracranial component is often intraluminal in the jugular vein. Vagal paraganglioma (glomus vagale) from the inferior ganglion are located between the skull base and hyoid bone, presenting commonly as a painless neck mass at the angle of the mandible. Paragangliomas only rarely secrete catecholamines. Therefore, preoperative 24-hour urine collection for norepinephrine and metabolites, including vanillymandelic acid, is not routinely performed for head and neck paraganglioma unless the patient has a suggestive history of palpitations, tachycardia, flushing, or excessive perspiration.
Parapharyngeal paragangliomas arise in the poststyloid compartment. CT and MRI have improved diagnosis of paragangliomas, with MRI venography and angiography adding to the definition of these tumors. Whereas carotid body paragangliomas are at the bifurcation of the internal and external carotid arteries, vagal paragangliomas displace the internal and external carotid arteries anteromedially and the internal jugular vein laterally. CT is best in determining bone involvement. MRI is superior in evaluating intracranial and vascular encroachment. Imaging in multiple planes is now possible with both CT and MRI. Paragangliomas exhibit a classic salt-and-pepper appearance on MRI because of hemorrhage and vascular flow voids, respectively.118
Paraganglia of the head and neck are neuroectoderm-derived chromaffin cells in extra-adrenal sites. Chromaffin cells secrete and store catecholamines. Paragangliomas have a high density of somatostatin type 2 receptors on the cell surface. Octreotide is a somatostatin analogue that when coupled to a radioisotope (indium 111) produces a scintigraphic image of these tumors both primary and recurrent.127 Noninvasive studies have replaced angiography as the primary diagnostic tool; however, presurgical angiographic evaluation and percutaneous transcatheter arterial embolization continue to play an important role in the management of these tumors in selected cases.128
Paraganglioma recapitulate the architecture of normal paraganglia. Paragangliomas are grossly firm, brown tumors with a pseudocapsule. Microscopically, they exhibit clusters of chief cells (type I, epithelioid cells) separated by a vascular stroma. The clustering pattern is known as zellballen (from the German meaning “cell balls”).129 The sustentacular cells (type II supporting cells) surround the edge of the cell balls (Fig. 9–19). Chief cells are immunoreactive to neuroendocrine markers, including synaptophysin, neuron-specific enolase, and chromogranin A.130 Sustentacular cells are immunoreactive to S-100 protein. Paragangliomas are similar histologically to adrenal pheochromocytoma.
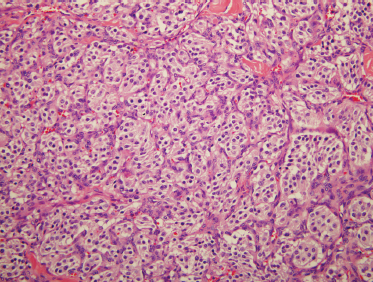
FIGURE 9-19 Histology of paraganglioma. These distinctive neoplasms contain nested neuroendocrine cells in balls (zellballen), surrounded by sustentacular cells (H&E, ×200).
Five percent of jugulotympanic and 10 to 15% of vagal paragangliomas are malignant.131 Malignant paragangliomas do not display nuclear and cellular pleomorphism and vascular or perineural invasion and are therefore not histologically distinguishable from benign paragangliomas.132 They are distinguished from benign tumors clinically by invasion and metastasis. Because paragangliomas can be multiple, metastasis is suggested when lesions are present at sites where paraganglioma tissue is not expected, such as cervical lymph nodes.
The most common genetic defect is the PGL1 gene at chromosome band 11q23. Genetic loci at PGL1, PGL2, and PGL3 are involved with familial transmission.133
Surgical management of paraganglioma is very challenging because multiple cranial nerves are often involved, with invasion of the skull base and intracranial extension. Observation with serial imaging studies must be presented to the patient. Focused radiation therapy can be used for selected high-risk tumors with expected postoperative multiple lower cranial nerve dysfunctions, recurrent tumors, bilateral tumors, incompletely resected tumors, or metastatic tumors. Paragangliomas often show no signs of progression after radiation therapy.134
If surgery is elected, an active skull base team consisting of a head and neck surgeon, vascular surgeon, neuro-otologist, neurosurgeon, neuroradiologist, and speech pathologist is necessary. A lateral approach starts with a level 2 and 3 selective neck dissection to improve access and rule out metastasis. The internal carotid artery, jugular vein, and CN IX, X, XI, and XII are identified. A small vagal paraganglioma may be removed using only a cervical approach. The tumor may be adherent to the carotid artery, and a bypass procedure may be required. Preoperative balloon occlusion test should be considered if the carotid artery appears at risk near the skull base. If the tumor approaches the jugular foramen, the jugular foramen may need to be exposed from a transmastoid approach. Removing the styloid process and associated musculature improves the exposure to separate the tumor from the carotid artery. With intracranial extension, superior control of the internal carotid artery deep to the glenoid fossa is necessary. Treatment of a dural defect can be managed with a fat graft or a superficial temporoparietal fascia flap.
Resection of these tumors generally requires sacrifice of the vagus nerve, but multiple lower cranial nerves can be impaired. Type I medialization thyroplasty can be performed at the time of surgery or in a delayed fashion. Delayed treatment allows the addition of the arytenoids adduction suture under local anesthesia and sedation where the responsive patient allows fine-tuning of this delicate procedure. Unilateral palatal adhesion can rehabilitate the palate in patients with velopharyngeal insufficiency.
 Neurofibromas
Neurofibromas
Neurofibromas can be asymptomatic and can rarely present in the parapharyngeal space as a solitary neck mass. They are also nerve sheath tumors (along with schwannomas) but in contrast to schwannomas are unencapsulated and grow between the nerve fibers of the parent nerve instead of pushing the nerve to one side. Histological exam reveals a spindle cell lesion with elongated, wavy nuclei. Solitary neurofibromas are best treated by complete surgical resection. In von Recklinghausen’s disease (neurofibromatosis) neurofibromas can be multiple and surgical treatment is pursued for symptomatic lesions. Von Recklinghausen’s disease is autosomal dominant with variable penetrance with half of patients having a family history and the other half resulting from spontaneous mutation. Initial clinical findings are café-au-lait spots and neurofibroma. Five or more brown cutaneous macules are diagnostic. Neurofibromas in von Recklinghausen’s disease mostly involve CN VIII. They can have sarcomatous transformation in von Recklinghausen’s disease in 10% of cases.135
 Traumatic Neuromas
Traumatic Neuromas
Traumatic neuromas may present as a small mass (×2cm) in the parotid or submandibular areas. They can be asymptomatic or present with paresthesia or tingling. Although they are most common after radical neck dissection, they can occur after salivary gland surgery or from blunt trauma to the neck. Traumatic neuromas are an attempt of an injured nerve to regenerate. They are fibrous masses with a mix of neural axons, Schwann’s cells, and perineural tissue. Excision is diagnostic, particularly in patients without a prior history of surgery.
 Other Benign Mesenchymal Tumors: Lipomas
Other Benign Mesenchymal Tumors: Lipomas
Lipomas are benign, often subcutaneous thinly encapsulated masses of adipose tissue. They occur infrequently in the salivary gland. They can occur in the superficial or deep lobe of the parotid as well as the parapharyngeal space. Lipomas have a wide age range, male predominance, slow growth rate, and are asymptomatic.136 The ultrasound appearance is hypoechoic. CT and MRI have characteristic appearances with ill-defined margins.137 Most lipomas of the parotid are composed of mature adipose tissue without myxoid, spindle cell, pleomorphic, or angiomatous features.55 Lipomas rarely recur after excision.
Sialolipomas have been described as a distinct salivary gland lipoma occurring in major and minor salivary glands. These are lipomas with secondary entrapment of salivary gland elements. Histologically, the tumors have a fibrous capsule of glandular tissue and mature adipose elements without atypia. There is more adipose component in the parotid tumors compared with the minor salivary gland tumors. The glandular components include ductal, acinar, basal, and myoepithelial cells resembling normal salivary gland structures. Their cell proliferative activity measured by Ki67 immunohistochemical staining is low, suggesting glandular components become entrapped during lipomatous proliferation rather than representing true neoplastic elements.136 Mixed tumors on rare occasion contain extensive fatty components and must be considered in the differential diagnosis. The fatty components in this type of mixed tumor may derive from pluripotential reserve cells of the mixed tumor.138 The sharp separation of the epithelial component from the adipose element and the presence of normal-appearing acinar cells characterize the sialolipoma and distinguish it from a mixed tumor.136 Superficial or partial superficial parotidectomy or resection of oral cavity tumor for the minor salivary gland sialolipoma is successful with rare recurrence.
Spindle cell lipomas are asymptomatic, slow-growing masses often 4 to 5 cm in size, occurring in males in their 60s and 70s. They must be distinguished from other spindle cell or mesenchymal tumors, including liposarcoma, schwannoma, leiomyoma, fibromatosis, melanoma, and malignant fibrous histiocytosis. They have mature fat cells, spindle cells in uniform arrangement, and areas of myxoid stromal change.139 Spindle cell lipoma is cured by simple excision and must be differentiated from more ominous lesions.
Hibernomas are benign lipomatous tumors developing from vestigial remnants of brown adipose cell that have remained from embryonic life.140 Other distinct microscopic variants of lipoma of the salivary glands include angiolipoma, fibrolipoma, and pleomorphic lipoma.136 Atypical lipomas are well-differentiated lipomatous lesions that are benign tumors in superficial tissue locations that can recur locally but do not metastasize and are distinguished from liposarcoma.
 Cysts of the Salivary Glands
Cysts of the Salivary Glands
About 5 to 10% of all salivary gland diseases are different types of salivary gland cysts (Table 9-4). Salivary gland cysts may be acquired or congenital. A true cyst has an epithelial lining in contrast to a pseudocyst as exemplified by a mucocele or a sialocele in the post-parotidectomy patient. Parotid cysts make up ×5% of parotid lesions. Pseudocysts are very common in minor salivary glands. Neoplasia with cystic changes must be excluded in salivary gland cystic lesions. Warthin’s tumor is the neoplasm most commonly associated with a cystic component. Benign mixed tumor, mucoepidermoid carcinoma, and adenoid cystic carcinoma are also associated with cyst formation. If fine-needle aspiration of a salivary gland lesion shows only cyst contents, repeat aspiration of any residual mass is important to exclude a neoplasm.
Table 9-4 Cystic Salivary Gland Lesions
| 1. Retention cysts |
| 2. Pseudocysts a. Traumatic-minor salivary gland b. Obstructive c. Ranula |
| 3. Chronic sialadenitis |
| 4. Benign lymphoepithelial cyst a. Non-HIV b. HIV |
| 5. Type I branchial cleft cyst |
| 6. Dermoid cyst |
| 7. Congenital polycystic parotid gland |
| 8. Lymphangioma |
| 9. Warthin’s tumor |
| 10. Cystic mixed tumor |
| 11. Cystadenoma |
| 12. Low-grade mucoepidermoid |
| 13. Adenoid cystic carcinoma |
| 14. Lymphoma |
HIV, human immunodeficiency virus
Acquired Cysts
A mucocele lacks an epithelial lining and is not a true cyst, and therefore is a pseudocyst. Mucoceles of the minor salivary gland are the most common salivary gland lesion. Mucoceles occur most commonly in the lips, particularly the lower lip, but also in the buccal mucosa and ventral tongue, during the first four decades of life. Mucoceles have a rapid onset, with fluctuation in size, bluish color, and fluid consistency. They have a preceding history of trauma (i. e., biting the lip) with rupture of the duct and mucous extravasation in the surrounding soft tissues and are surrounded by granulation tissue. Type IV collagenases and plasminogen activators are proteolytic enzymes thought to participate in the formation of mucoceles.141 Mucoceles contain polymorphonuclear leukocytes and macrophages. Complete excision will prevent recurrence. Mucoceles of the anterior lingual salivary glands (glands of Blandin and Nuhn) can persist if glands of the deep tongue musculature are not excised.142 Cryosurgery is an alternative management strategy for mucoceles.143
Minor salivary gland duct obstruction can result in a mucous retention cyst or sialocyst, a true cyst with an epithelial lining. Retention cysts are less common than mucoceles and do not often have an antecedent history of trauma. They may occur from a partial obstruction of the duct.55 In the minor salivary glands, they most commonly occur in the lips, but also in the buccal mucosa and ventral tongue. Most patients with minor salivary gland retention cysts are in their fourth decade of life. Complete excision will prevent recurrence.
Retention cysts in major salivary glands are called salivary duct cysts or sialocysts. The cause of the duct obstruction in these epithelial-lined cysts is not always known. They are generally slow-growing, painless lesions. They are usually unilocular, but can be multilocular. Retention cysts are treated with complete excision to prevent recurrence.
Epithelial alterations in salivary duct cysts of the parotid gland and in mucous retention cysts of minor salivary glands include metaplasias (goblet cells, clear cells, squamous cells) and focal epithelial proliferations. These changes are comparable to similar alterations in odontogenic cysts. Retention cysts or salivary duct cysts may be the starting point of a salivary gland tumor such as a cystadenoma or mucoepidermoid carcinoma in rare cases.144
Benign Lymphoepithelial Cysts
Benign lymphoepithelial cysts in non-HIV patients form from epithelial ductular inclusions in lymph nodes that then become cystic.145 They form mostly unilaterally, but occasionally bilaterally, predominantly in adults, in their fourth and fifth decade of life, as painless, asymptomatic masses. Prior to the AIDS epidemic, they were typically identified in the floor of the mouth and less commonly the parotid.
They have a similar histology whether from the major or minor salivary glands. Histological exam shows a squamous epithelial-lined unilocular cyst within or associated with lymph nodes. Multilocular presentation is rare. Fine-needle aspiration of the cyst contents show a population of lymphocytes, histiocytes, plasma cells, and metaplastic squamous cells in a proteinaceous background.15 It is important to distinguish between lymphoepithelial cysts and various lymphoid lesions, including reactive lymph nodes and lymphoma. Other neoplastic processes with a significant lymphoid component include Warthin’s tumor, sebaceous lymphadenoma, and lymphoepithelial carcinoma. Benign lymphoepithelial cysts associated with HIV and those seen in benign lymphoepithelial lesion (or myoepithelial sialadenitis) of Sjögren’s are discussed in Chapter 5. Treatment of benign lymphoepithelial cysts depends on symptoms and the suspicion of malignancy.
Branchial Cleft Cysts and Ranulas
See Chapter 8
Sialadenosis
Sialadenosis or sialosis is a noninflammatory, nonneoplastic, mostly symmetric enlargement of the salivary glands, sometimes associated with pain. The parotid glands are most commonly involved, but structural and functional changes can occur in the submandibular and minor salivary glands.146 The enlargement is gradual, bilateral, but not always symmetric. There is no gender predilection. The peak incidence is between 30 and 70 years of age.147
An underlying systemic cause for sialadenosis is almost always found. Three broad headings (Table 9-5) for the cause of sialadenosis are endocrine (diabetes mellitus, adrenal disorders), dystrophic-metabolic (alcoholism and malnutrition), and neurogenic (anticholinergic medications disrupting the autonomic nervous system).148 In contrast, Mikulicz’s disease is an historic term that describes patients with asymptomatic enlargement of the salivary glands or lacrimal glands without an underlying disease. Sialadenosis must be distinguished from other causes of bilateral parotid swelling including Sjoögren’s syndrome, sialadenitis, sarcoidosis, lymphoma, and tuberculosis.
Parotid enlargement has been described in nearly all endocrine disorders. Diabetes is the most common, but it is also observed in hyperthyroidism, hypothyroidism, pancreatitis, pregnancy, lactation, puberty, menopause, and testicular and ovarian atrophy.55
Table 9-5 Causes of Sialadenosis
Endocrine
|
Dystrophic-metabolic
|
Neurogenic
|
Alcoholic cirrhosis is associated with sialadenosis in up to 50% of cases. Sialadenosis is very rare in nonalcoholic cirrhosis. In chronic alcoholics, modification in acinar cells is noted; however, the most evident changes occur in the ductal system, where enlargement is present. Other findings include atrophy of epithelial cells and desquamated cells.146
Malnutrition leading to sialadenosis can occur in kwashiorkor, hypovitaminosis, beriberi, pellagra, anorexia, and bulimia. Any disease that interferes with absorption of nutrients can lead to sialadenosis, including Chagas’ disease, celiac disease, and bacillary dysentery. Up to half of the patients with bulimia will exhibit sialadenosis. Malnutrition in bulimic patients results in acinar enlargement of the salivary glands with plump pyramidal cells containing prominent zymogen granules.148
Salivary gland hypertrophy is associated with medicines including phenothiazine, phenobarbitol, iodine-containing products, isoproterenol, ethambutol, and heavy metals, some that affect the autonomic nervous system.
Sialography in the later stages of the disease demonstrates compression of the proximal ducts by acinar swelling. Early stages of the disease may show no sialographic changes.148
Normal acinar cells are 30 to 40 mm, whereas in sialadenosis, the diameters are enlarged to 50 to 70 mm.149 Acinar cell enlargement and disturbance of secretion in sialadenosis are pathologic findings hypothesized to occur from a peripheral autonomic neuropathy, a demyelinating polyneuropathy.147 Ultrastuctural support for this notion comes from Donath and Seifert,149 who have demonstrated degenerative changes both in myoepithelial cells and in the postganglionic sympathetic neurons of the autonomic nervous system, including loss of neurosecretory granules, destruction of mitochondria, hydropic swelling of the axoplasm, and a terminal axolysis. These cellular changes may result in a lengthening of the storage phase of the secretory granules and an arrest in protein synthesis by the acinar cells, both processes producing accumulation of granules and marked enlargement of acini.150
Three histological patterns of sialadenosis are recognized: granular, honeycomb or vacuolar transformation, and mixed.149, 150 Ultrastructural evidence reveals that acinar cells in sialadenosis are filled with zymogen granules of three parallel varieties. There is a dark granule type with densely packed protein-rich granules, but no evidence of increased protein secretion. A light granule type contains low optical density secreting granules and increased synthesis of protein. A mixed granule type has light and dark acinar cells.149 Fatty infiltration or lipomatosis and fibrosis of the interlobular septae are additional histological findings, along with naked nuclei. Inflammatory infiltrates are not seen.
Sialadenosis can be confused with a tumor, and a biopsy is diagnostic. This can often be accomplished with fine-needle aspiration where normal salivary gland acini and ducts are found. Careful correlation with radiology is essential to make sure that the finding of normal salivary gland tissue is representative of the process and is not merely a sampling error missing a discrete mass.151 Amylase levels in the saliva and serum are commonly increased, assisting in the diagnosis. A careful history, however, may obviate the need for a biopsy.
Correction of the underlying systemic problem usually results in reduction in the size of the gland. Salivary substitutes, heat application to the swollen salivary glands, and sialagogues can be helpful. Pilocarpine, a cholinergic-mimetic medication, can reduce the parotid gland swelling in the bulimic patient.152 The salivary gland swelling can be resistant to correction of the underlying problem. Refractory cases are often secondary to an endocrine and neurogenic basis.148 Surgery is rarely indicated in refractory cases to improve unacceptable appearance.
 Other Causes of Salivary Gland Enlargement
Other Causes of Salivary Gland Enlargement
Bilateral parotid enlargement is seen in obesity because of fatty infiltration. Obesity is associated with diabetes mellitus and therefore may be caused by sialadenosis. Dehydration with salivary stasis and mucous plugging of salivary ducts result in swelling and pain. This enlargement is readily treated with rehydration, massage of the gland, and sialagogues. Pneumoparotitis can occur after intubation or endoscopy and can be seen in glass blowers.
Sclerosing polycystic adenosis is a rarely reported entity described as a pseudoneoplastic reactive inflammatory process of the major salivary glands similar to sclerosing adenosis of the breast.153 Patients present at a mean age of 30 and have a female predominance. They develop a slow-growing unilateral mass in the parotid (less often in the submandibular gland) with pain or a tingling sensation. These lesions consist of incompletely encapsulated circumscribed masses characterized by hyalinized and sclerotic collagenous tissue with cystically dilated ducts and focal epithelial hyperplasia.153,154 Treatment is excision with adequate margins. Recurrences are possible.
Enlargement of the minor salivary glands of the lips in adults with a clear, sticky mucous is called cheilitis glandularis. Excretory duct dilation results in eversion and enlargement of the lip. Three classifications based on clinical and histological findings have been described: simple, superficial, and deep.
Adenomatoid hyperplasia of the minor salivary glands is a rare idiopathic condition resulting in a painless, firm mass covered by intact oral mucosa usually of the hard and soft palate. They occur in the fourth to sixth decade more commonly in male patients. These well-circumscribed, unencapsulated lesions form lobules of normal-appearing mucinous acini within the lamina propria and submucosa. The treatment is excision.155
Accessory parotid gland tissue occurs anterior to the parotid in the distribution of the buccal nerve branches of the facial nerve. Heterotopic salivary gland tissue can be identified most commonly in periparotid and intra-parotid lymph nodes. The neck at the anterior border of the sternocleidomastoid muscle in the area of the sternoclavicular joint is also an area of salivary gland heterotopia. More unusual locations include the pituitary, mandible, maxilla, lower neck, larynx, hypopharynx, middle ear, thyroglossal duct, mediastinum, prostrate, vulva, stomach, and rectum.15 Their presumed origin is salivary gland rests. Generally, they are an asymptomatic incidental finding, although they can present as a mass or fistula. Heterotopic salivary gland tissue is treated with simple excision, and neoplasms of heterotopic salivary tissue are treated by the pathology of the tumor.
REFERENCES
1. Everson, JW Cawson, R. Salivary gland tumours: a review of 2410 cases with particular reference to histological types, site, age and sex distribution. J Pathol 1985; 146: 51–58
2. Spiro, R. Salivary neoplasms: overview of a 35-year experience with 2,807 patients. Head Neck Surg 1986; 8: 177–184
3. Foote, FW Frazell, L. Tumors of the major salivary glands. Cancer 1953; 6: 1065–1074
4. Ahlbom, H. Mucous and salivary gland tumours: a clinical study with special reference to radiotherapy based on 254 cases treated at Radiumhemmet, Stockholm. Acta Radiol Suppl 1935; 23: 1–452
5. Stein, I Geschickter, CF. Tumors of the parotid gland. Arch Surg 1934; 28: 482–526
6. Carwardine, T. Excision of the parotid gland with preservation of the facial nerve. Lancet 1907; ii: 892
7. Sistrunk, W. Mixed tumor of the parotid gland. Minn Med 1921; 4: 155–160
8. McFarland, J. Three hundred mixed tumors of the salivary glands of which 69 recurred. Surg Gynecol Obstet 1936; 63
9. Patey, DH Thackray, AC. The treatment of parotid tumours in the light of a pathological study of parotidectomy material. Br J Surg 1957; 45: 477–487
10. Smiddy, FG. Treatment of mixed parotid tumours. Br Med J 1956; 1: 322–325
11. Lack, E Upton, M. Histopathologic review of salivary gland tumors in childhood. Arch Otolaryngol Head Neck Surg 1988; 114: 898–906
12. Toh, H Kodama, J Fukuda, J, et al. Incidence and histology of human accessory glands. Anat Rec 1993; 236: 589–590
13. Gnepp, D. Schroeder W, Heffner D. Synchronous tumors arising in a single major salivary gland. Cancer 1989; 63: 1219–1224
14. Alves, FA Perez, DE Almeida, OP Lopes, MA Kowalski, LP. Pleomorphic adenoma of the submandibular gland: clinicopathological and immunohistochemical features of 60 cases. Arch Otolaryngol Head Neck Surg 2002; 128: 1400–1403
15. Gnepp, D Brandwein, M Henley, J. Salivary and lacrimal glands. In: Gnepp, D, ed. Diagnostic surgical pathology of the head and neck. Philadelphia: WB Saunders; 2001: 325–430
16. Chao, P Lee, F. Pleomorphic adenoma (chondroid syringoma) on the face. Otolaryngol Head Neck Surg 2004; 130: 499–500
17. Bialek, EJ Jakubowski, W Karpinska, G. Role of ultrasonography in diagnosis and differentiation of pleomorphic adenomas: work in progress. Arch Otolaryngol Head Neck Surg 2003; 129: 929–933
18. Shah, GV. MR imaging of salivary glands. Magn Reson Imaging Clin N Am 2002; 10: 631–632
19. Keyes, J. Salivary gland tumors: pretherapy evaluation with PET. Radiology 1994; 192: 99–103
20. Dardick, I van Nostrand, AW. Myoepithelial cells in salivary gland tumors revisited. Head Neck Surg 1985; 7: 395–408
21. Dardick, I van Hostrand, AW Phillips, MJ. Histogenesis of salivary gland pleomorphic adenoma (mixed tumor) with an evaluation of the role of the myoepithelial cell. Hum Pathol 1982; 13: 62–75
22. Batsakis, J Regezi, J. The pathology of head and neck tumors: salivary glands. Head Neck Surg 1978; 1: 59–68
23. Seifert, G Langrock, I Donath, K. [Pathologic and morphologic subclassification of salivary pleomorphic adenoma.] HNO 1976; 24: 415–426
24. Ellis, G Auclair, P. Benign epithelial neoplasms. In: Ellis, G Auclair, P, eds. Atlas of Tumor Pathology: Tumors of the Salivary Glands. Washington, DC: Armed Forces Institute of Pathology; 1995: 39–153
25. Hubner, G Kleinsasser, O Klein, H. The fine structure of salivary duct carcinomas: on the role of myoepithelial cells in tumors of the salivary glands [in German]. Virchows Arch A Pathol Pathol Anat 1969; 346: 1–14
26. Naeim, F Forsberg, MI Waisman, J Coulson, WF. Mixed tumors of the salivary glands: growth pattern and recurrence. Arch Pathol Lab Med 1976; 100: 271–275
27. Stennert, E Guntinas-Lichius, O Klussmann, J Arnold, G. Histopathology of pleomorphic adenoma in the parotid gland: a prospective unselected series of 100 cases. Laryngoscope 2001; 111: 2195–2200
28. Voz, M Van de Ven, W Kas, K. First insight into the molecular basis of pleomorphic adenomas of the salivary glands [review]. Adv Dent Res 2000; 14: 81–83
29. Enlund, F Nordkvist, A Sahlin, P Mark, J Stenman, G. Expression of PLAG1 and HMGIC proteins and fusion transcripts in radiation-associated pleomorphic adenomas. Int J Oncol 2002; 20: 713–716
30. Tsuzuki, H Saito, H Imamura, Y Noriki, S Fukuda, M. Early progression stage of malignancy as revealed by immunohistochemical demonstration of DNA instability: 2. Otorhinolaryngeal border-line neoplastic lesions. Eur J Histochem 1994; 38: 291–302
31. Witt, R. The significance of the margin in parotid surgery for pleomorphic adenoma. Laryngoscope 2002; 112: 2141–2154
32. Sungur, N Akan, I Ulusoy, M Ozdemir, R Kilinc, H Ortak, T. Clinicopathological evaluation of parotid gland tumors: a retrospective study. J Craniofac Surg 2002; 13: 26–30
33. Laccourreye, H Laccourreye, O Cauchois, R, et al. Total conservative parotidectomy for primary benign pleomorphic adenoma of the parotid gland: a 25-year experience with 229 patients. Laryngoscope 1994; 104: 1487–1494
34. McGurk, M Renehan, A Gleave, E Hancock, B. Clinical significance of the tumour capsule in the treatment of parotid pleomorphic adenomas. Br J Surg 1996; 83: 1747–1749
35. Lam, K Wei, W Ho, H Ho, C. Whole organ sectioning of mixed parotid tumors. Am J Surg 1990; 160: 377–381
36. Carew, JF Spiro, RH Singh, B Shah, JP. Treatment of recurrent pleomorphic adenomas of the parotid gland. Otolaryngol Head Neck Surg 1999; 121: 539–542
37. Stennert, E Wittekindt, C Klussmann, J Arnold, G Guntinas- Lichius, O. Recurrent pleomorphic adenoma of the parotid gland: a prospective histopathological and immunohistochemical study. Laryngoscope 2004; 114: 158–163
38. Glas, A Vermey, A Hooema, H, et al. Surgical treatment of recurrent pleomorphic adenoma of the parotid gland: a clinical analysis of 52 patients. Head Neck 2001; 23: 311–316
39. Niparko, J Beauchamp, M Krause, C Baker, S Work, W. Surgical treatment of recurrent pleomorphic adenoma of the parotid gland. Arch Otolaryngol Head Neck Surg 1986; 112: 1180–1184
40. Myssiorek, D Ruah, C Hybels, R. Recurrent pleomorphic adenomas of the parotid gland. Head Neck 1990; 12: 332–336
41. Fee, W Goffinet, D Calcaterra, T. Recurrent mixed tumors of the parotid gland: results of surgical therapy. Laryngoscope 1978; 88: 265–273
42. Maran, AG Mackenzie, IJ Stanley, RE. Recurrent pleomorphic adenoma of the parotid gland. Arch Otolaryngol 1984; 110: 167–171
43. Witt, R. Facial nerve function after partial superficial parotidecotomy: an 11-year review (1987–1997). Otolaryngol Head Neck Surg 1999; 121: 210–213
44. Gleave, E Whittaker, J Nicholson, A. Salivary tumours: experience over thirty years. Clin Otolaryngol 1979; 4: 247–257
45. Conley, J. Problems with reoperation of the parotid gland and facial nerve. Otolaryngol Head Neck Surg 1988; 99: 480–488
46. Maynard, J. Enucleated parotid tumours. Br J Surg 1988; 75: 764–766
47. Junquera, L Alonso, D Sampedro, A Fresno, F Albertos, J Lopez- Arranz, J. Pleomorphic adenoma of the salivary glands: prospective clinicopathologic and flow cytometric study. Head Neck 1999; 21: 652–656
48. Bankamp, D Bierhoff, E. Proliferative activity in recurrent and nonrecurrent pleomorphic adenoma of the salivary glands. Laryngorhinootologie 1999; 78: 77–80
49. Glas, A Hollema, H Nap, R Plukker, J. Expression of extrogen receptor, progesterone receptor, and insulin-like growth factor receptor-1 and of MIB-1 in patients with recurrent pleomorphic adenoma of the parotid gland. Cancer 2002; 94: 2211–2216
50. Niparko, J Kileny, P Kemink, J Lee, H Graham, M. Neurophysiologic intraoperative monitoring: 2. Facial nerve function. Am J Otol 1989; 10: 55–61
51. Renehan, A Gleave, EN McGurk, M. An analysis of the treatment of 114 patients with recurrent pleomorphic adenomas of the parotid gland. Am J Surg 1996; 172: 710–714
52. Douglas, J Einck, J Austin-Seymour, M Koh, W Laramore, G. Neutron radiotherapy for recurrent pleomorphic adenomas of major salivary glands. Head Neck 2001; 23: 1037–1042
53. Hanna, D Dickison, W Richardson, G, et al. Management of recurrent salivary gland tumors. Am J Surg 1976; 132: 453–458
54. Patel, N Poole, A. Recurrent benign parotid tumours: the lesson not learnt yet? Aust N Z J Surg 1998; 68: 562–564
55. Ellis, G Auclair, P. Tumors of the salivary gland. In: Ellis, G Auclair, P, eds. Atlas of Tumor Pathology. Washington, DC: Armed Forces Institute of Pathology; 1996: 39–153
56. Barnes, L Appel, B Perez, H El-Attar, A. Myoepithelioma of the head and neck: case report and review. J Surg Oncol 1985; 28: 21–28
57. el-Naggar, A Callender, D Ordonez, N Killary, A. Cytogenetic analysis of a primary salivary gland myoepithelioma. Cancer Genet Cytogenet 1999; 113: 49–53
58. Dardick, I Van Nostrand, A. Myoepithelial cells in salivary gland tumors revisited. Head Neck Surg 1985; 7: 395–408
59. Sciubba, J Brannon, R. Myoepithelioma of the salivary glands: a case report of 23 cases. Cancer 1982; 49: 562–572
60. Dardick, I Thomas, M van Nostrand, A. Myoepithelioma—new concepts of histology and classification: a light and electron microscopic study. Ultrastruct Pathol 1989; 13: 187–224
61. Gnepp, D Brandwein, M Henley, J. Salivary and lacrimal glands. In: Gnepp, D, ed. Diagnostic Surgical Pathology of the Head and Neck. Philadelphia: WB Saunders; 2001: 325–430
62. McCluggage, W Primrose, W Toner, P. Myoepithelial carcinoma (malignant myoepithelioma) of the parotid gland arising in a pleomorphic adenoma. J Clin Pathol 1998; 51: 552–556
63. Warthin, A. Papillary cystadenoma lymphomatosum. J Cancer Res 1929; 13: 116–125
64. Hildebrand, O. [Innate epithelial cysts and growths of the ncek.] Arch F Klin Chir 1895; 49: 167–192
65. Vories, A Ramirez, S. Warthin’s tumor and cigarette smoking. South Med J 1997; 90: 416–418
66. Maiorano, E LoMuzio, L Favia, G Piattelli, A. Warthin’s tumour: a study of 78 cases with emphasis on bilaterality, multifocality and association with other malignancies. Oral Oncol 2002; 38: 35–40
67. Seifert, G Donath, K. Multiple tumours of the salivary glands: terminology and nomenclature. Eur J Cancer B Oral Oncol 1996; 32B:3–7
68. Ellies, M Laskawi, R Arglebe, C. Extraglandular Warthin’s tumours: clinical evaluation and long-term follow-up. Br J Oral Maxillofac Surg 1998; 36: 52–53
69. Yoo, G Eisele, D Askin, F Driben, J Johns, M. Warthin’s tumor: a 40-year experience at The Johns Hopkins Hospital. Laryngoscope 1994; 104: 799–803
70. Hill, A. Major salivary gland tumours in a rural Kenyan hospital. East Afr Med J 2002; 79: 8–10
71. Saku, T Hayashi, Y Takahara, O, et al. Salivary gland tumors among atomic bomb survivors, 1950–1987. Cancer 1997; 79: 1465–1475
72. Sato, T Morita, Y Hamamoto, S, et al. Interpretation of scintigraphy of papillary cystadenoma lymphomatosum (Warthin’s tumor) on the basis of histopathologic findings. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1996; 82: 101–107
73. Ikarashi, F Nakano, Y Nonomura, N Kawana, M. Radiological findings of adenolymphoma (Warthin’s tumor). Auris Nasus Larynx 1997; 24: 405–409
74. Horiuchi, M Yasudi, S Shohtsu, A Ide, M. Four cases of Warthin’s tumor of the parotid gland detected with FDG PET. Ann Nucl Med 1998; 12: 47–50
75. Thompson, A Bryant, H. Histogenesis of papillary cystadenoma lymphomatosum (Warthin’s tumor) of the parotid salivary gland. Am J Pathol 1950; 26: 807–849
76. Eveson, J Terry, J Alfonso, A. Warthin’s tumor (cystadenolymphoma) of salivary glands: a clinicopathologic investigation of 278 cases. Oral Surg Oral Med Oral Pathol 1986; 61: 256–262
77. Gallo, O Bocciolini, C. Warthin’s tumour associated with autoimmune diseases and tobacco use. Acta Otolaryngol 1997; 117: 623–627
78. Aguirre, JM Echebarria, MA Martinez-Conde, R Rodriguez, C Burgos, JJ Rivera, JM. Warthin’s tumor: a new hypothesis concerning its development. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1998; 85: 60–63
79. Ellies, M Laskawi, R Arglebe, C. Extraglandular Warthin’s tumours: clinical evaluation and long-term follow-up. Br J Oral Maxillofac Surg 1998; 36: 52–53
80. Santucci, M Gallo, O Calzolari, A Bondi, R. Detection of Epstein- Barr viral genome in tumor cells of Warthin’s tumor of parotid gland. Am J Clin Pathol 1993; 100: 662–665
81. Ogata, T Hongfang, Y Kayano, T Hirai, K. No significant role of Epstein-Barr virus in the tumorigenesis of Warthin’s tumor. J Med Dent Sci 1997; 44: 45–52
82. Brennan, PA Umar, T Smith, GI McCauley, P Peters, WJ Langdon, JD. Expression of type 2 nitric oxide synthase and p53 in Warthin’s tumor of the parotid. J Oral Pathol Med 2002; 31: 458–462
83. Thackray, A Lucas, R. Tumors of the major salivary glands. In: Atlas of Tumor Pathology. (2nd series). Washington, DC: Armed Forces Institute of Pathology; 1974: Fascicle 10
84. Shin, H Sneige, N. Sources of diagnostic error in fine needle aspiration diagnosis of Warthin’s tumor and clues to a correct diagnosis. Diagn Cytopathol 1997; 17: 230–234
85. Lewis, P Baxter, P Griffiths, A Parry, J Skibinski, D. Detection of damage to the mitochondrial genome in the oncocytic cells of Warthin’s tumor. J Pathol 2000; 191: 274–281
86. Shintaku, M Honda, T. Identification of oncocytic lesions of salivary glands by anti-mitochondrial immunohistochemistry. Histopathology 1997; 31: 408–411
87. Di Palma, S Simpson, RH Skalova, A Michal, M. Metaplastic (infracted) Warthin’s tumour of the parotid gland: a possible consequence of fine needle aspiration biopsy. Histopathology 1999; 35: 432–438
88. Seifert, G Bull, H Donath, K. Histologic subclassification of the cystadenolymphoma of the parotid gland: analysis of 275 cases. Virchows Arch A Pathol Anat Histol 1980; 388: 13–38
89. Schwerer, MJ Kraft, K Baczako, K Maier, H. Cytokeratin expression and epithelial differentiation in Warthin’s tumor and it metaplastic (infracted) variant. Histopathology 2001; 39: 347–352
90. Martins, C Fonseca, I Roque, L Soares, J. Cytogenetic characterization of Warthin’s tumour. Oral Oncol 1997; 33: 344–347
91. Parwani, A Ali, S. Diagnostic accuracy and pitfalls in fine-needle aspiration interpretation of Warthin’s tumor. Cancer 2003; 99: 166–171
92. Batori, M Mariotta, G Giovannone, G Casella, G Casella, M. Warthin’s tumor of parotid gland: treatment of a retroneural lesion by enucleation. Eur Rev Med Pharmacol Sci 2002; 6: 105–111
93. Leverstein, H Van der Wal, J Tiwari, R Van der Waal, I Snow, G. Results of the surgical management and histopathological evaluation of 88 parotid gland Warthin’s tumours. Clin Otolaryngol 1997; 22: 500–503
94. Warnock, G. Papillary cystadenoma lymphomatosum (Warthin’s tumor). In: Ellis, G Auclair, A Gnepp, D, eds. Surgical Pathology of the Salivary Gland. Philadelphia: WB Saunders; 1991: 187–201
95. Chhieng, DC Paulino, AF. Basaloid tumors of the salivary glands. Ann Diagn Pathol 2002; 6: 364–372
96. Stanley, MW Horwitz, CA Rollins, SD, et al. Basal cell (monomorphic) and minimally pleomorphic adenomas of the salivary glands: distinction from the solid (anaplastic) type of adenoid cystic carcinoma in fine-needle aspiration. Am J Clin Pathol 1996; 106: 35–41
97. Yu, G Ubmuller, J Donath, K. Membranous basal cell adenoma of the salivary gland: a clinicopathologic study of 12 cases. Acta Otolaryngol 1998; 118: 588–593
98. Choi, H Batsakis, J Callender, D Prieto, V Luna, M el-Naggar, A. Molecular analysis of chromosome 16q regions in dermal analogue tumors of salivary glands: a genetic link to dermal cylindroma? Am J Surg Pathol 2002; 26: 778–783
99. Machado de Sousa, S Soares de Araujo, N Correa, L Pires Soubhia, A Cavalcanti de Araujo, V. Immunohistochemical aspects of basal cell adenoma and canalicular adenoma of salivary glands. Oral Oncol 2001; 37: 365–368
100. Harmse, J Saleh, H Odutoye, T Alsanjari, N Mountain, R. Recurrent canalicular adenoma of the minor salivary glands in the upper lip. J Laryngol Otol 1997; 111: 985–987
101. Ferreiro, JA. Immunohistochemical analysis of salivary gland canalicular adenoma. Oral Surg Oral Med Oral Pathol 1994; 78: 761–765
102. Gnepp, D Schroeder, W Heffner, D. Synchronous tumors arising in a single major salivary gland. Cancer 1989; 63: 1219–1224
103. Ellis, G Auclair, P. Atlas of tumor pathology. In: Ellis, G Auclair, P, ed. Tumors of the Salivary Glands. Washington, DC: Armed Forces Institute of Pathology; 1996: 103, 318–114, 324
104. Ito, K Tsukuda, M Kawabe, R, et al. Benign and malignant oncocytoma of the salivary glands with an immunohistochemical evaluation of Ki-67. ORL J Otorhinolaryngol Relat Spec 2000; 62: 338–341
105. Davy, C Dardick, I Hammond, E Thomas, M. Relationship of clear cell oncocytoma to mitochondrial-rich (typical) oncocytomas of parotid salivary gland: an ultrastructural study. Oral Surg Oral Med Oral Pathol 1994; 77: 469–479
106. Ellis, G. Clear cell neoplasms in salivary glands: clearly a diagnostic challenge [review]. Ann Diagn Pathol 1998; 2: 61–78
107. Fantasia, JE Miller, AS. Papillary cystadenoma lymphomatosum arising in minor salivary glands. Oral Surg Oral Med Oral Pathol 1981; 52: 411–416
108. Damm, D White, D Geissler, R, et al. Benign solid oncocytoma of intraoral minor salivary gland. Oral Surg Oral Med Oral Pathol 1989; 67: 84–86
109. Martin, H Janda, J Behrbohm, H. [Locally invasive oncocytoma of the paranasal sinuses.] Zentralbl Allg Pathol 1990; 136: 703–706
110. Ishikawa, T Imada, S Ijuhin, N. Intraductal papilloma of the anterior lingul salivary gland: case report and immunohistochemical study. Int J Oral Maxillofac Surg 1993; 22: 116–117
111. Maiorano, E Favia, G Ricco, R. Sialadenoma papilliferum: an immunohistochemical study of five cases. J Oral Pathol Med 1996; 25: 336–342
112. Brannon, RB Sciubba, JJ Guilani, M. Ductal papillomas of salivary gland origin: a report of 19 cases and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2001; 92: 68–77
113. Fantasia, JE Nocco, CE Lally, ET. Ultrastucture of sialadenoma papilliferum. Arch Pathol Lab Med 1986; 110: 523–527
114. Linhartova, A. Sebaceous glands in salivary gland tissue. Arch Pathol 1974; 98: 320–324
115. Maruyama, S Cheng, J Inoue, T Takagi, R Saku, T. Sebaceous lymphadenoma of the lip: report of a case of minor salivary gland origin. J Oral Pathol Med 2002; 31: 242–243
116. Rosner, M Fisher, W Mulligan, L. Cervical sympathetic schwannoma. Neurosurgery 2001; 49: 1452–1454
117. Wilson, JA McLaren, K McIntyre, MA von Haacke, NP Maran, AG. Nerve-sheath tumors of the head and neck. Ear Nose Throat J 1988; 67: 103–110
118. Hamza, A Fagan, J Weissman, J Myers, E. Neurilemomas of the parapharyngeal space. Arch Otolaryngol Head Neck Surg 1997; 123: 622–626
119. Jager, L Reiser, M. CT and MR imaging of the normal and pathologic conditions of the facial nerve. Eur J Radiol 2001; 40: 133–146
120. Maly, B Maly, A Doviner, V Reinhartz, T Sherman, Y. Fine needle aspiration biopsy of intraparotid schwannoma. Acta Cytol 2003; 47: 1131–1134
121. Bretlau, P Melchiors, H Krogdahl, A. Intraparotid neurilemmoma. Acta Otolaryngol 1983; 95: 382–384
122. Manolidis, S Shohet, J Jackson, C, et al. Malignant glomus tumors. Laryngoscope 1999; 109: 30–34
123. Wasserman, P Savargaonkar, P. Paragangliomas classification, pathology and differential diagnosis. Otolaryngol Clin North Am 2001; 34: 845–862
124. Balatsouras, D Eliopoulos, P Economou, C. Multiple glomus tumours. J Laryngol Otol 1992; 106: 538–543
125. Maier, W Marangos, N Laszig, R. Paraganglioma as a systemic syndrome: pitfalls and strategies. J Laryngol Otol 1999; 113: 978–982
126. Coia, LR Fazekas, JT Farb, SN. Familial chemodectoma. Int J Radiat Oncol Biol Phys 1981; 7: 949–952
127. Bustillo, A. Octreotide scintigraphy in the detection of recurrent paragangliomas. Otolaryngol Head Neck Surg 2004; 130: 479–482
128. Rao, AB Koeller, KK Adair, CF. Paragangliomas of the head and neck: radiologic-pathologic correlation. Radiographics 1999; 19: 1605–1632
129. Barnes, L. Paragangliomas of the larynx. ORL J Otorhinolaryngol Relat Spec 1991; 53: 220–234
130. Ferlito, A Friedmann, I. Contribution of immunohistochemistry in the diagnosis of neuroendocrine neoplasms of the larynx. ORL J Otorhinolarynol Relat Spec 1991; 53: 235–244
131. Kahn, L. Vagal body tumor with cervical lymph node metastasis and familial association: ultrastructural study and review. Cancer 1976; 38: 2367–2377
132. Urquhart, A Johnson, J Myers, E, et al. Glomus vagale: paragangliomas of the vagus nerve. Laryngoscope 1994; 104: 440–445
133. Baysal, B. Genetics of familial paragangliomas. Otolaryngol Clin North Am 2001; 34: 863–879
134. Mendenhall, W Parsons, J Stringer, S, et al. Radiotherapy in the management of temporal bone chemodectoma. Skull Base Surg 1995; 5: 83–91
135. Ducatman, B Scheithauer, BW Piepgras, DG Reiman, HM Ilstrup, DM. Malignant peripheral nerve sheath tumors: a clinicopathologic study of 120 cases. Cancer 1986; 57: 2006–2021
136. Nagao, T Sugano, I Ishida, Y, et al. Sialolipoma: a report of seven cases of new variant of salivary gland lipoma. Histopathology 2001; 38: 30–36
137. Chikui, T Yonetsu, K Yoshiura, K, et al. Imaging findings of lipomas in the orofacial region with CT, US, and MRI. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1997; 84: 88–95
138. Seifert, G Donath, K Schafer, R. Lipomatous pleomorphic adenoma of the parotid gland: classification of lipomatous tissue in salivary glands. Pathol Res Pract 1999; 195: 247–252
139. Fletcher, C Martin-Bates, E. Spindle cell lipoma: a clinicopathological study with some original observation. Histopathology 1987; 11: 803–817
140. Vinayak, BC Reddy, KT. Hibernoma in the parotid region. J Laryngol Otol. 1993; 107: 257–258
141. Azuma, M Tamatani, T Fukui, K, et al. Proteolytic enzymes in salivary extravasation mucoceles. J Oral Pathol Med 1995; 24: 299–302
142. Sugerman, PB Savage, NW Young, WG. Mucocele of the anterior lingual salivary glands (glands of Blandin and Nuhn): report of 5 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2000; 90: 478–482
143. Toida, M Ishimaru, J Hobo, N. A simple cryosurgical method for treatment of oral mucous cysts. Int J Oral Maxillofac Surg 1993; 22: 353–355
144. Seifert, G. Mucoepidermoid carcinoma in a salivary duct cyst of the parotid gland: contribution to the development of tumours in salivary gland cysts. Pathol Res Pract 1996; 192: 1211–1217
145. Bernier, J Bhaskar, S. Lymphoepithelial lesions of salivary glands: histogenesis and classification based on 186 cases. Cancer 1958; 11: 1165–1179
146. Ferraris, M Arriaga, A Busso, C Carranza, M. Histological study of parotid, submaxillary and von Ebner salivary glands in chronic alcoholics. Acta Odontol Latinoam 1999; 12: 97–102
147. Kim, D Uy, C Mandel, L. Sialosis of unknown origin. N Y State Dent J 1998; 64: 38–40
148. Coleman, H Altini, M Nayler, S Richards, A. Sialadenosis: a presenting sign in bulimia. Head Neck 1998; 20: 758–762
149. Donath, K Seifert, G. Ultrastructural studies of the parotid glands in sialadenosis. Virchows Arch A Pathol Anat Histol 1975; 365: 119–135A
150. Seifert, G Miehlke, J Haubrich, J Chilla, R. Diseases of the salivary glands: pathology-diagnosis-treatment facial nerve surgery. Stuttgart: Georg Thieme Verlag; 1986: 78–84
151. Henry-Stanley, M Beneke, J Bardales, R Stanley, M. Fine-needle aspiration of normal tissue from enlarged salivary glands: sialosis or missed target? Diagn Cytopathol 1995; 13: 300–303
152. Mehler, PS Wallace, JA. Sialadenosis in bulimia. Arch Otolaryngol Head Neck Surg 1993; 119: 787–788
153. Smith, B Ellis, G Slater, L Foss, R. Sclerosing polycystic adenosis of major salivary glands: a clinicopathologic analysis of nine cases. Am J Surg Pathol 1996; 20: 161–170
154. Mackle, T Mulligan, M Dervan, P O’Dwyer, T. Sclerosing polycystic sialadenopathy. Arch Otolaryngol Head Neck Surg 2004; 130: 357–360
155. Buchner, A Merrell, PW Carpenter, WM Leider, AS. Adenomatoid hyperplasia of minor salivary glands. Oral Surg Oral Med Oral Pathol 1991; 71: 583–587
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


