CHAPTER 9 Antimuscarinic Drugs
Various drugs can interfere with the transmission of nerve impulses at cholinergic junctions. As shown in Table 5-2, some drugs prevent the uptake of choline by the nerve terminal or the release of acetylcholine (ACh) from the terminal; other drugs block at ganglia or, by a competitive or depolarizing form of blockade, at neuromuscular junctions. The drugs presented in this chapter block responses in muscarinic receptors and are essentially without effect except at inordinately high doses at nicotinic receptors. These drugs are known as antimuscarinic or muscarinic receptor–blocking drugs; the term anticholinergic, although often used for this class of drugs, is inaccurate because these drugs, for the most part, are selective for muscarinic receptors but not nicotinic receptors. They are also termed atropine-like because of their derivation from or relation to the oldest and best-known member of the group. Because peripheral muscarinic receptors are the primary targets of ACh released by postganglionic cholinergic neurons, the effects achieved by the antimuscarinic drugs are chiefly on smooth muscle, cardiac muscle, and glands that are innervated by these neurons.
CHEMISTRY AND CLASSIFICATION
Antimuscarinic drugs fall into four categories, as follows:
A prototypic chemical structure of each of these types is shown in Table 9-1, and a more extensive list is at the end of the chapter.
TABLE 9-1 Chemical Structures of Representatives of the Four Classes of Antimuscarinic Drugs
| TYPE OF COMPOUND | EXAMPLE | CHEMICAL STRUCTURE |
|---|---|---|
| Naturally occurring alkaloid | Atropine | 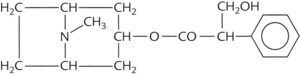 |
| Semisynthetic derivative of alkaloid | Methscopolamine | 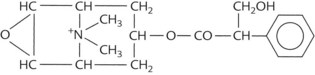 |
| Synthetic quaternary ammonium compound | Propantheline | 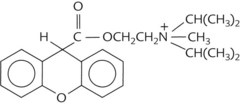 |
| Synthetic but not quaternary ammonium compound | Benztropine | 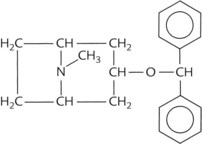 |
MECHANISM OF ACTION
The antimuscarinic drugs—whether the naturally occurring alkaloids or the semisynthetic or synthetic derivatives—are competitive antagonists of ACh at muscarinic receptors. (Review Figure 5-1 for the principal location of muscarinic receptors.) They have an affinity for muscarinic receptor sites but lack intrinsic activity.1 They occupy the receptor sites and prevent access of ACh, creating a blockade that is generally reversible by increasing the amount of ACh in the area of the receptor, as would occur after the administration of an anticholinesterase drug. Because atropine can antagonize the muscarinic effects of the anticholinesterases and vice versa, each drug can be used as an antidote for the other in case of poisoning. The antimuscarinic drugs are capable of blocking responses to parasympathetic nerve stimulation, to sympathetic nerve stimulation of thermoregulatory sweat glands, to ACh protected from hydrolysis by anticholinesterases, and to direct-acting muscarinic agents, although their capability for inhibiting the latter two is greater than for the first two.
Several explanations have been offered for why atropine is more effective in blocking the pharmacologic effects produced by muscarinic receptor agonists than in blocking physiologic responses evoked by parasympathetic nervous system stimulation. One possibility is that ACh released into the restricted environs of a junctional cleft may overwhelm the antagonist by the high, although temporary, concentrations achieved. A second possibility is that the antimuscarinic drugs facilitate ACh release from cholinergic neurons by blocking presynaptic muscarinic receptors that limit evoked ACh release. A third explanation arises from the fact that physiologic responses to parasympathetic nervous system stimulation are mediated by several neurotransmitters in addition to ACh. Direct electrical stimulation of the parasympathetic nervous system causes the release of ACh and several other neurotransmitters from the postganglionic nerve terminal.7 ACh and adenosine triphosphate (ATP) are released from postganglionic parasympathetic nerves. In this setting, ATP, acting on nucleotide receptors, functions as a cotransmitter with ACh.9
Although atropine is a highly effective antagonist at all muscarinic receptors, evidence has accumulated that there are five muscarinic subtypes, M1 to M5, each with different affinities for certain muscarinic agonists and antagonists, different anatomic distributions, and different second messenger signaling mechanisms (see Chapters 5 and 8). The relatively selective affinity of the tricyclic benzodiazepine pirenzepine for M1 receptors versus M2 and M3 receptors gives it stronger antimuscarinic properties in certain sites (e.g., corpus striatum, cerebral cortex, and enterochromaffin cells) compared with others (e.g., heart and ileum). Pirenzepine, which is available outside of the United States, was the first clinically useful selective muscarinic receptor antagonist. Darifenacin is a selective antagonist at the M3 receptor and is available for treatment of overactive bladder.14 The characterization of different muscarinic receptor subtypes continues to provide an impetus for development of selective antagonists.
PHARMACOLOGIC EFFECTS
Therapeutic doses of antimuscarinic drugs produce effects attributable to the blockade of peripheral muscarinic receptors and similar receptors in the central nervous system (CNS) located within the medulla and higher cerebral centers. In the following discussion, atropine and scopolamine, which have always been considered the prototypes for this class of drugs, are principally reviewed, but (1) atropine and scopolamine differ in the relative intensity of their antimuscarinic effects on specific organs (Table 9-2); (2) there is a difference in the susceptibility of various effectors to antimuscarinic agents in general (Table 9-3); (3) because of differences in chemical structure, some antimuscarinic drugs pass readily into the CNS, whereas others do not; (4) there are some major differences among antimuscarinic drugs in the onset and duration of their actions (Table 9-4); and (5) muscarinic receptor subtypes have differing affinities for specific antimuscarinic drugs.

TABLE 9-3 Order of Susceptibility of Effectors to Increasing Doses of Antimuscarinic Agents
| RESPONSE | DOSE |
|---|---|
| Secretion (saliva, sweat, bronchial) | Low |
| Mydriasis, cycloplegia, tachycardia | ↓ |
| Loss of parasympathetic control of urinary bladder and gastrointestinal smooth muscle | |
| Inhibition of gastric secretion | High |
TABLE 9-4 Onset and Duration of Cycloplegia Induced by Some Topical Antimuscarinic Drugs
| DRUG | ONSET (min) | DURATION |
|---|---|---|
| Atropine | 30-40 | ≥6 days |
| Scopolamine | 20-30 | 3-6 days |
| Homatropine | 40-60 | 1-3 days |
| Cyclopentolate | 25-75 | 6-24 hr |
| Tropicamide | 20-35 | 2-6 hr |
Peripheral Nervous System Actions
The antimuscarinic drugs possess peripheral nervous system and CNS actions, but the nature and intensity of these vary with the individual drug and the dose administered. Most peripheral nervous system effects are caused by an interruption of parasympathetic impulses to a given effector. This interruption results in control of the tissue or organ by the sympathetic nervous system, which often exerts effects opposite to those of the parasympathetic nervous system. An important exception is where the sympathetic effect acts through muscarinic receptors, most notably in the sweat glands. The sympathetic effect of sweating is inhibited by antimuscarinic drugs. The pharmacologic effects observed depend largely on the existing activity of postganglionic cholinergic neurons. Inhibition of sweating and hyperthermia are likely to be observed on a hot day, but no effect on thermoregulation is apparent in a cold environment. Generally, atropine-like drugs block the salivation, lacrimation, urination, and defecation response to cholinergic drugs described in Chapter 8 and the hypotensive and bradycardic effects of muscarinic receptor stimulation. The effects of antimuscarinic agents on specific tissues are described next.
The eye
Atropine-like drugs block muscarinic receptors in the sphincter of the iris and in the ciliary muscle, leading to dilation of the pupil (mydriasis) and paralysis of accommodation (cycloplegia). Photophobia and fixation of the lens occurs for far vision, and vision for near objects is blurred. Intraocular pressure is not significantly affected except in the case of narrow-angle (or angle-closure) glaucoma, for which administration of these drugs may cause a dangerous increase in intraocular pressure. The onset and duration of the mydriatic and cycloplegic effects differ, as shown for cycloplegia in Table 9-4, and to some extent the choice of an agent for an ophthalmologic procedure is influenced by these differences.
Salivary glands
Parasympathetically mediated salivary secretion is abolished in a dose-dependent manner, whereas salivary gland vasodilation is much less affected. The mouth and throat become unpleasantly dry, to the point that speech and swallowing may become difficult. Dry mouth or xerostomia can lead to numerous adverse effects on the oral cavity (see Chapter 8).
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


