CHAPTER 41 Immunotherapy
Immunopharmacology is the study of the interaction between drugs and the immune system. Immunotherapy is the application of clinical strategies to modulate the activities of certain components of the immune system in order to improve immune function and to prevent or treat disease. This chapter reviews the immune system with regard to the pathways used toward adaptive, specific immunity that are or can be targeted for immunotherapy and discusses immunotherapeutic strategies that are of clinical importance today or show promise for the future. The pharmacologic manipulation of innate immune mechanisms involved in inflammation is covered in Chapters 21 and 35.
OVERVIEW OF SPECIFIC IMMUNITY
Components of the Immune System
Cells
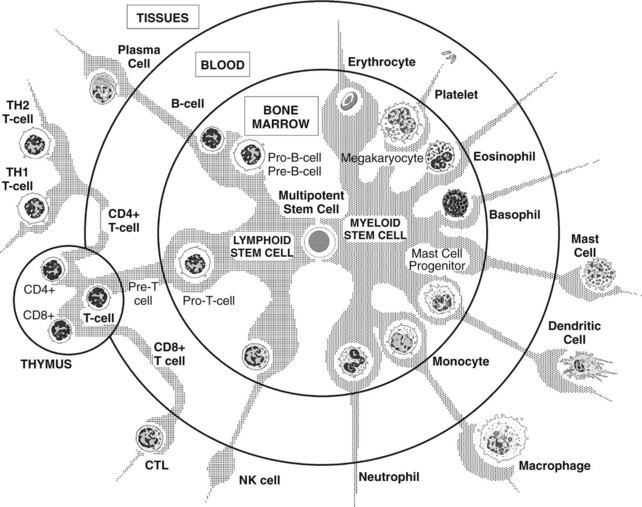
All the effectors of the immune system are derived from the bone marrow. Pluripotent hematopoietic stem cells in bone marrow give rise to either myeloid progenitor cells or lymphoid progenitor cells (Figure 41-1). Myeloid progenitor cells are the precursors of red blood cells, platelets, granulocytes (polymorphonuclear leukocytes [PMNs]: neutrophils, eosinophils, and basophils), monocyte-macrophages, dendritic cells (DCs), and mast cells. Lymphoid progenitor cells give rise to the T, B, and NK cells. Lymphocytes are generated and mature in the bone marrow and thymus, which are considered to be the primary or central lymphoid organs. From there, they travel to reside in secondary or peripheral lymphoid organs, such as lymph nodes, spleen, mucosa-associated lymphoid tissues (MALT), gut-associated lymphoid tissues (GALT), and bronchus-associated lymphoid tissues (BALT).
Receptors and other cell-surface proteins
Mediators
Cytokines
An important feature of cytokine action is that multiple cytokines often work in concert to foster a particular change in cellular activity. The proliferation and differentiation of effector T cells important in cell-mediated immunity (CMI) depend on the interplay of TH1 cytokines such as IL-2, IL-12, and IFN-γ. Activation of B cells for humoral immunity is based on the release of several interleukins (IL-4, IL-5, IL-10, and IL-13) by TH2 cells. TH1 cytokines are important in stimulating B-cell differentiation leading to the production of immunoglobulins IgG1, IgG2, and IgG3. TH2 cytokines stimulate IgE and IgG4 production. The actions of selected cytokines are summarized in Table 41-1.
TABLE 41-1 Selected Cytokines and Their Functional Relationships
| CYTOKINE | SECRETED BY | FUNCTIONS |
|---|---|---|
| Lymphoid Hematopoiesis | ||
| IL-7 | SC |
This summary table is not intended to provide a complete listing of all the biologic functions of the cytokines, but rather to point out the relationships among them.
Ag, Antigen; APC, antigen-presenting cell; B, B cell; CMI, cell-mediated immunity; CSF, colony-stimulating factor; En, endothelial cell; Ep, epithelial cell; Fb, fibroblast; G, granulocyte; L, lymphoid cell; M, monocyte; Ma, macrophage; My, myeloid cell; OAF, osteoclast-activating factor; RC, renal cortex; SC, stromal cell; T, T cell; TNF, tumor necrosis factor.
Humoral antibodies
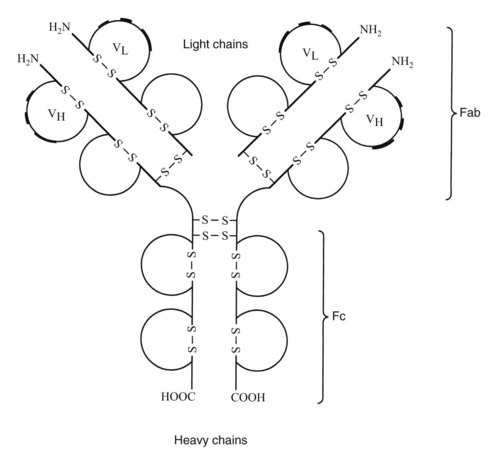
Antibodies synthesized and released by plasma cells directly mediate humoral immunity. As shown in Figure 41-2, the basic immunoglobulin structure consists of two heavy chains and two light chains covalently linked by interchain disulfide bonds. Both chains consist of two or more domains, each defined by a single intrachain disulfide bond. The heavy chain is composed of three or four constant domains and one variable domain. The light chain incorporates one constant and one variable domain. The relatively flexible hinge regions found in certain immunoglobulins are believed to be remnants of primordial constant domains. Terminal sequences on the amino end of each chain make up the variable regions of the molecule. Within each variable region are hypervariable sequences that are responsible for specific antigen binding. There are two types of light chains, λ and κ. The ratio of λ and κ chains differs in various species, being 1 : 20 in mice and 1 : 2 in humans. Sometimes the ratio of λ to κ is used to identify multiple myeloma.
Initiation, Progression, and Termination of Specific Immune Responses
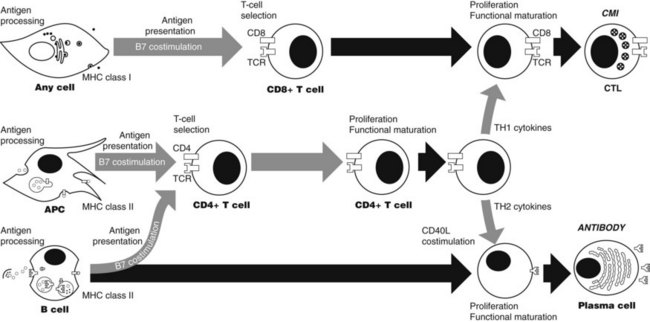
Specific immune responses involve a series of events (Figure 41-3), each of which offers a potential site for immunotherapeutic intervention. Included in this series are antigen processing and presentation, T-cell selection, lymphocyte differentiation, effector function, and termination. These events occur in response to changes in the concentrations of antigens intracellularly and extracellularly.
T-cell activation
The actual recognition of antigen by the TCR is a low-affinity reaction. This characteristic enables many TCRs of a given T cell to interact with the few specific antigens presented by the antigen-presenting cell. Multiple interactions are important because T-cell activation depends on the number of TCRs that interact with antigen over time.42 The factors that influence T-cell activation include the number of antigen molecules presented by the antigen-presenting cell, the affinity of the TCR for the antigen, and the number of TCRs. If the interaction with peptide antigen by TCRs is sufficient, the TCRs cluster on the T-cell surface and downregulate (the TCRs are probably internalized). With costimulation, downregulation of approximately 4250 TCRs leads to T-cell activation.
Differentiation
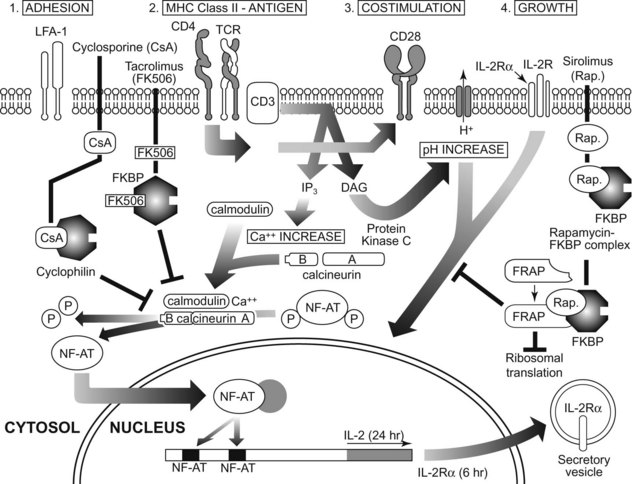
After antigen recognition by TCR and BCR, intracellular signaling events allow the T cell to differentiate into functionally mature cells (Figure 41-4). Much present-day immunotherapy is aimed at this stage of the specific immune response. Binding of antigen to its receptor leads to the clustering of antigen receptor on lymphocytes, which is the first step in signal transduction, Clustering of antigen receptor leads to activation of intracellular signaling molecules. Protein tyrosine kinases are enzymes that affect the function of other proteins by adding a phosphate group to certain tyrosine residues. Specific growth factors such as c-kit have cytoplasmic domains that contain intrinsic tyrosine kinase activity. T- and B-cell signaling is different from c-kit signaling since the TCR and BCR do not have intrinsic kinase domains; rather they rely on the interaction with other tyrosine kinases known as receptor-associated tyrosine kinases.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


