CHAPTER 8 Cholinergic Drugs
Cholinergic drugs are agents that mimic the actions of the endogenous neurotransmitter acetylcholine (ACh). As described in Chapter 5, ACh is the primary neurotransmitter released from the nerve terminals of the preganglionic fibers of the parasympathetic and sympathetic nervous systems, the postganglionic fibers of the parasympathetic nervous system (which include most of the postganglionic cholinergic neurons), and some postganglionic fibers of the sympathetic nervous system (mostly fibers to the sweat glands). ACh is also the primary neurotransmitter released from somatic efferents innervating skeletal muscle and from certain central nervous system (CNS) neurons.
CHOLINOMIMETIC AGONISTS
Chemistry and Classification
Choline esters
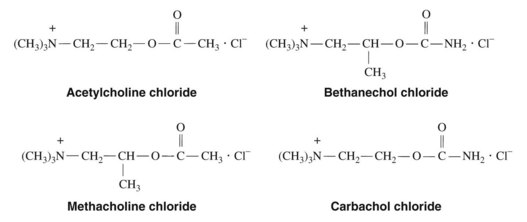
The history of the discovery of ACh and its identification is described in Chapter 5. In 1909, Hunt synthesized the acetyl ester of choline, and earlier Hunt and Taveau16 reported on the pharmacology of many synthetic congeners of ACh. Interest in the choline esters arose partly out of the hope that some of these compounds would have a longer duration of action than ACh and, at the same time, a greater degree of selectivity. This goal has not been realized completely, and ACh and related drugs generally either are not used therapeutically or are used only in selected instances. The structures of ACh and the three principal synthetic esters of choline—methacholine, carbachol, and bethanechol—are shown in Figure 8-1. Succinylcholine, a diacetylcholine derivative with selective nicotinic receptor effects in skeletal muscle, is discussed in Chapter 10.
Natural alkaloids and congeners
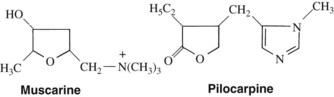
Several alkaloids obtained from various plants possess direct cholinomimetic activity. Muscarine, the prototype muscarinic agonist, is present during certain times of the year in the mushroom Amanita muscaria and is especially prominent in several Inocybe and Clitocybe species. Although a quaternary ammonium compound (Figure 8-2), muscarine has a rapid onset of action after oral ingestion and produces physiologic responses characteristic of profound parasympathetic nervous system stimulation. In severe poisoning, cardiovascular collapse may occur. Pilocarpine is found in the leaves of the South American shrub Pilocarpus jaborandi. It is also a selective muscarinic receptor agonist. Pilocarpine remains in the therapeutic armamentarium for a few specific indications and has a specific dental indication. Cevimeline, a synthetic agent, is similar in pharmacology to pilocarpine. Arecoline is the primary alkaloid of betel nuts. It is a euphoretic and stimulates muscarinic and ganglionic nicotinic receptors. Nicotine, an alkaloid found in tobacco leaves (Nicotiana tabacum), is important historically as the prototype nicotinic receptor agonist. In the form of cigarettes, nicotine is the most commonly used cholinergic agonist, and it is responsible for the physical dependence associated with smoking. This drug and other drugs selective for nicotinic receptors are discussed in Chapter 10.
Mechanism of Action
Parasympathomimetic responses to cholinergic drugs are mediated by the stimulation of several populations of muscarinic receptors. A total of five muscarinic receptor proteins (m1 through m5, corresponding to the pharmacologically identified receptors M1 through M5) have been produced from cloned muscarinic receptor genes, and it has been established that multiple receptor subtypes can coexist in the same organ or tissue. The exact distribution of these receptors and their functional properties are currently areas of active investigation, but a few general concepts have emerged. In the periphery, the M1 receptor seems to be localized in ganglia, some exocrine gland cells, and the enterochromaffin cells of the stomach (see Chapter 33). The M2 receptor is the primary subtype found in the heart and is present, along with the M4 receptor, in the lung. The M3 receptor is widely distributed and is most prominent in glandular tissue. Although a peripheral distribution of the M5 receptor has not been identified, it is expressed, as are the other subtypes, in discrete regions of the CNS.
Muscarinic receptors belong to a large family of plasma membrane receptors whose basic structure consists of seven helical segments spanning the membrane and joined by alternating intracellular and extracellular peptide bridges (see Chapter 1). Although the hydrophobic helical segments, which form the ligand-binding site, show considerable structural homology among the muscarinic receptor subtypes, the third intracellular loop, joining helices V and VI, is highly divergent. Biochemical studies suggest that this loop is of primary importance in the coupling between receptor binding and intracellular action.
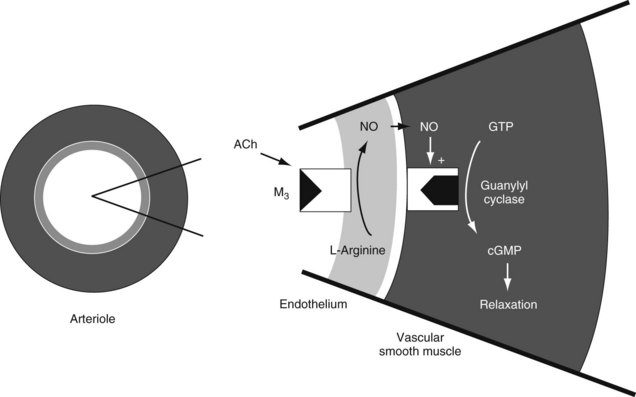
Stimulation of muscarinic receptors initiates a cascade of intracellular events that ultimately leads to the observed pharmacologic effects. Evidence to date suggests that all muscarinic receptor subtypes regulate the activity of G proteins (see Chapter 5). The G proteins modulate intracellular processes by influencing “second messenger” systems. Agonist-induced activation of M1, M3, or M5 receptors stimulates the enzyme phospholipase C, which produces Ca++-dependent phosphorylation of specific cellular regulatory proteins. The stimulation of M2 or M4 receptors inhibits the activity of adenylyl cyclase, decreasing the intracellular concentration of cyclic adenosine 3′,5′-monophosphate. In the heart, this outcome of M2 receptor activation results in increased K+ efflux and reduced Ca++ influx, leading to characteristic muscarinic receptor–induced changes in cardiac function (see later). The activation of M2 receptors on the intact vascular endothelium produces a profound vasodilation by stimulating the production and release of nitric oxide, an important endothelium-derived relaxing factor (Figure 8-3).13,17 Nitric oxide stimulates guanylyl cyclase located in vascular smooth muscle, which catalyzes the formation of cyclic guanosine 3′,5′-monophosphate. This cyclic nucleotide reduces intracellular Ca++ concentrations, leading to vascular smooth muscle relaxation and vasodilation. The effect of agonists on the muscarinic receptors of endothelial cells accounts for the vasodilation when these drugs are administered systemically, especially intravenously. This vasodilation occurs despite the lack of nerve innervation to these receptors on endothelial cells.
The systemic administration of high doses of ACh activates nicotinic receptors located on the cell bodies of postganglionic nerve fibers of the autonomic nervous system (NN receptors) and nicotinic receptors located in the neuromuscular junction (NM receptors). As described in Chapter 5, nicotinic receptors are composed of five glycoprotein subunits forming a rosette around a central channel spanning the plasma membrane. The α subunits (see also Figure 1-2) contain the ACh-binding sites. When stimulated by ACh, nicotine, or another nicotinic receptor agonist, a conformational change in the protein occurs, allowing Na+ and, to a lesser extent, Ca++ ions to move down their respective concentration gradients. The net ionic movement depolarizes the postganglionic cell body or muscle end plate. Prolonged stimulation of nicotinic receptors with ACh or nicotine results in a phenomenon referred to as “depolarization blockade,” in which responses to further stimulation are attenuated and then lost (see Chapter 10).
Pharmacologic Effects
Peripheral muscarinic effects
Cholinergic agonists that stimulate muscarinic receptors produce end-organ responses that mimic parasympathetic nervous system stimulation. Table 5-1 outlines several of the physiologic responses produced by direct electric stimulation of parasympathetic nerves. The following discussion of the specific muscarinic effects of the cholinergic drugs is limited to actions that have some therapeutic application or toxicologic importance; not all of the cholinergic drugs possess all these actions.
Vascular smooth muscle
Muscarinic receptor agonists produce a generalized vasodilation that causes a decrease in blood pressure. All vascular beds are affected, which is consistent with pharmacologic evidence that all parts of the vasculature, including pulpal blood vessels, are supplied with muscarinic receptors. The physiologic significance of these receptors is still in doubt, however, partly because much of the vasculature receives no parasympathetic innervation. In the absence of an administered drug, it is likely that vasodilation in local tissues occurs most often in response to autoregulatory factors, such as high carbon dioxide concentrations, low oxygen concentrations, and an acidic pH, and not to stimulation of cholinergic nerves. ACh produced and released locally may facilitate vasodilation in response to local blood flow increases. As noted previously, muscarinic receptor agonists produce their vasodilatory effects by inducing the vascular endothelium to release nitric oxide into the surrounding vascular smooth muscle, where it produces muscle relaxation.13,17
Bronchial smooth muscle
The smooth muscle of the bronchioles is constricted by muscarinic receptor agonists.
Peripheral nicotinic effects
Several cholinomimetic drugs can stimulate nicotinic receptors. Nicotinic receptor agonists have varying effects at different nicotinic sites; these effects are related to the structure of the molecule,3 the dosage of the drug, and the location and type of nicotinic receptor activated. As noted earlier, there are at least two major kinds of peripheral nicotinic receptors: those on ganglia (NN) and those in skeletal muscle (NM). Although exogenous ACh at low doses stimulates muscarinic receptors selectively, in substantially higher doses it stimulates NN receptors and, by close intra-arterial injection of high doses, NM receptors. Carbachol has substantial nicotinic properties at therapeutic doses. Its affinity for nicotinic receptors is higher than that for muscarinic receptors. There is evidence that carbachol not only occupies the postsynaptic cholinergic receptor but also causes the release of ACh from nerve terminals in certain locations by activating presynaptic nicotinic receptors. Muscarinic effects are obtained indirectly through increased ACh release at parasympathetic ganglia and muscarinic neuroeffector sites. Although pilocarpine is essentially muscarinic in action, it has been reported to produce ganglionic stimulation in high doses.
Stimulation of autonomic ganglia leads to a mixture of parasympathetic and sympathetic effects. Because these effects often oppose each other, the resultant outcome is often difficult to predict. In the case of ACh and carbachol, which also exert prominent muscarinic activity, parasympathetic effects predominate; this is likely due to the fact that these drugs have a more difficult time gaining access to nicotinic receptors. The pharmacology of nicotine, which is devoid of direct muscarinic properties, is reviewed in Chapter 10. None of these agents produces clinically useful skeletal muscle stimulation.
Adverse Effects
The mushrooms Amanita pantherina and Amanita muscaria contain muscarine but in amounts that are probably too small to account for the symptoms of poisoning that result from their ingestion. The mushroom Inocybe lateraria, with a much higher muscarine content, produces signs and symptoms of intoxication that resemble those produced by muscarine, including profuse salivation and sweating; miosis; bradycardia; severe abdominal pain with vomiting, cramps, and diarrhea; and respiratory difficulties arising from the constriction of bronchial muscle and increased secretion in the respiratory tract. The onset of poisoning is rapid, and treatment consists of the administration of atropine in large quantities, gastric lavage, and appropriate supportive measures. Recovery usually occurs in 1 or 2 days. In many cases of mushroom poisoning, there are delayed symptoms, including violent emesis and diarrhea and damage to parenchymatous organs (principally the liver), which are not amenable to atropine treatment and are produced by a group of cyclopeptide toxins from the mushroom that inhibit the synthesis of messenger ribonucleic acid.2
ANTICHOLINESTERASES
Physostigmine, or eserine, the earliest known anticholinesterase, has a colorful history. An alkaloid, it is derived from a bean, or nut, known as the Calabar, ordeal, or Esére bean, and it was used in witchcraft trials by certain native tribes in West Africa. The bean was brought to England by a British medical officer stationed in Calabar in the mid-1800s, and its pharmacologic properties were investigated in numerous laboratories, including those of Fraser, who studied its toxicity in the 1860s and noted that its actions were antagonized by atropine. In 1877, physostigmine was used for the treatment of glaucoma, which remains one of its principal uses today. In 1914, noting the extreme brevity of the action of ACh, Dale8 suggested that an enzyme capable of destroying ACh must exist in the body, and in 1930 it was found that physostigmine could prevent the rapid destruction of ACh.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


