CHAPTER 7 Adrenergic Antagonists
Understanding of the mechanisms of transmission in the sympathetic nervous system has increased significantly as a result of a better understanding of the actions of drugs; an increased appreciation of receptors and the second messenger pathways used by them; and extensive investigation of important diseases such as congestive heart failure, coronary artery disease, and hypertension. The consequence of this work has been the development of numerous new pharmacologic agents possessing increasingly selective mechanisms of action. Chapter 5 discusses the theoretic mechanisms by which drugs produce effects on the autonomic nervous system (see Table 5-2). The drugs discussed in this chapter all interfere with sympathetic nervous system transmission. Despite having diverse mechanisms of action, these drugs are collectively referred to as adrenergic antagonists or sympatholytics. Most, but not all, adrenergic antagonists are competitive antagonists of either α-adrenergic or β-adrenergic receptors (adrenoceptors). As a result, these agents block the actions of the endogenous neurotransmitters epinephrine and norepinephrine and exogenously administered adrenergic agonists and are also called adrenergic receptor blockers.
A group of agents with sympatholytic activity includes drugs that are agonists at α2-adrenergic receptors in key brain nuclei controlling cardiovascular function (see Chapter 6). These drugs reduce blood pressure largely by decreasing the outflow of sympathetic nervous system transmission to cardiovascular effectors. Several agents that are mainly of historical interest are known collectively as adrenergic neuron–blocking drugs. They act on nerve terminals to produce their sympatholytic effects and are discussed in Chapter 28.
HISTORY
Evidence that drugs could be used to antagonize the actions of other pharmacologic agents was obtained shortly after the isolation and synthesis of epinephrine. In 1906, Dale noticed that certain alkaloids isolated from ergot, produced by a fungus disease of rye grain, blocked the ability of epinephrine to increase systemic arterial blood pressure. After an injection of ergotoxine (a mixture of ergot alkaloids), a hypotensive effect was observed in response to epinephrine treatment; it was aptly named by Dale the “epinephrine reversal” response.9 These early studies also provided the first example of selective antagonism by showing that ergot derivatives were capable of blocking some, but not all, of the actions of epinephrine. This idea of selective antagonism remains an important aspect of drug development and use.
The pioneering work of Ahlquist1 in delineating the α-adrenergic and β-adrenergic receptors provided the framework necessary to classify more systematically antagonists of sympathetic nervous system function. Nickerson and Goodman21 reported in 1947 the development of dibenamine, an agent capable of irreversibly blocking the α-adrenergic receptor, which inhibited certain responses to exogenous epinephrine and to adrenergic nerve stimulation. Selective blockade of α-adrenergic receptors by ergotoxine also explained the epinephrine reversal described by Dale.9 Phentolamine and related imidazolines were early examples of nonselective, competitive antagonists at α-adrenergic receptors. Selective antagonists of the α1-adrenergic and α2-adrenergic receptors have now been developed. Dichloroisoproterenol was the first β-adrenergic receptor blocker developed. The first clinically useful β blocker introduced was propranolol, which blocks β1-adrenergic and β2-adrenergic receptors. Selective β1 antagonists were then discovered. We now know that there are at least nine adrenergic receptors (α1A, α1B, α1D, α2A, α2B, α2C, β1, β2, and β3). Increasingly selective antagonists against each of these receptors are being developed with the goal of obtaining drugs capable of specifically interfering with the receptor involved in a pathophysiologic condition without blockade of other receptors that could lead to unwanted side effects.
In addition to effects on adrenergic receptors, it was found that sympathetic nervous system transmission could also be altered by actions directly on the nerve terminal. The drug bretylium interferes with the release of norepinephrine in response to nerve stimulation.4 Other drugs were also developed that interfere with neuronal function at the level of the nerve terminal. Reserpine depletes the norepinephrine stored in these nerve terminals.5 Metyrosine (α-methyl-l-tyrosine) competitively inhibits tyrosine hydroxylase, the rate limiting enzyme in the synthesis of norepinephrine.11 These sympatholytics are not as therapeutically beneficial as the selective receptor antagonists, however, because their administration is associated with many unpleasant side effects. In the 21st century, the use of these agents has been curtailed, and they have been largely relegated to historical footnotes. Brief descriptions of these drugs are given in Chapters 5, 12, and 28.
SELECTIVE α1-ADRENERGIC RECEPTOR ANTAGONISTS
As discussed in Chapter 5, α1-adrenergic receptors are located predominantly on the postjunctional membranes of glands and smooth muscle. The α1-adrenergic receptors associated with smooth muscle of arteries and veins play an important role in promoting vasoconstriction and in regulating systemic arterial blood pressure and blood flow. The α1-adrenergic receptors are also important in regulating the tone of nonvascular smooth muscle, such as in the neck of the urinary bladder and capsule of the prostate. More recent evidence has suggested that the α1-adrenergic receptor plays a role in the regulation of hypertrophic growth, the generation of reactive oxygen species, and apoptotic cell death. Antagonism of these cellular events may also be the reason that α1-adrenergic receptor blockers are effective in the treatment of benign prostatic hyperplasia. The α2-adrenergic receptors are found on the prejunctional neuronal membrane, where they play an autoregulatory role in inhibiting norepinephrine release. They are also located postjunctionally on the membranes of pancreatic islet cells, smooth muscle, and blood platelets.17
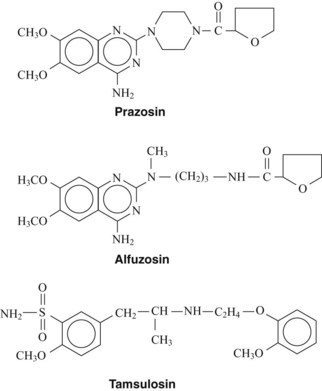
Prazosin and Analogues
The first therapeutically useful α1-adrenergic receptor antagonist developed was prazosin (Figure 7-1).3 Terazosin and doxazosin are structural analogues that were subsequently introduced. Although these agents differ in pharmacokinetic properties, their mechanism of action is the same. The α1-adrenergic receptor antagonists prevent the action of sympathetic neurotransmitters and exogenously administered agonists at α1-adrenergic receptors on effector organs. Prazosin and related compounds have essentially equal affinity for all three subtypes (α1A, α1B, and α1D) of the α1-adrenergic receptor.
As a result of blocking smooth muscle α1-adrenergic receptors, prazosin dilates arterioles and veins. Each of these actions contributes to the hypotension seen with this drug. Blockade of arterial smooth muscle produces hypotension by reducing peripheral resistance. The venodilation resulting from blocked venous α1-adrenergic receptors decreases cardiac preload. Compared with the nonselective α receptor antagonists, prazosin causes less tachycardia, a smaller increase in cardiac output, and less renin release.3
Therapeutic uses
Prazosin, terazosin, and doxazosin can be used in monotherapy for the treatment of hypertension (see Chapter 28). Terazosin and doxazosin, which are given once a day, may have advantages over prazosin, which requires more frequent administration. Otherwise, the clinical effects of terazosin and doxazosin are similar to the effects of prazosin. Although prazosin and analogues can alleviate the signs and symptoms of congestive heart failure (because of a reduction in preload and afterload), they have not been shown to increase survival in patients with congestive heart failure.20,23 The doxazosin arm of a more recent clinical trial, the ALLHAT study, was discontinued because of increased cardiovascular risks compared with alternative treatments.20 This finding has resulted in a significant reduction in the use of prazosin analogues in the therapy of hypertension. These drugs do not have adverse effects on lipids or cholesterol and may be particularly useful in treating patients with hyperlipidemia.7,18 Prazosin and its analogues are also effective in treating benign prostatic hyperplasia caused by the blocking of the α1-adrenergic receptors associated with smooth muscle of the bladder neck and prostate. This action reduces pressure on the urethra and improves urine flow. Because of their longer plasma half-lives, terazosin and doxazosin may be preferred over prazosin for this indication.7,18
Alfuzosin
Alfuzosin (see Figure 7-1) is an analogue of prazosin that selectively blocks the α1-adrenergic receptors associated with the prostate gland.19 Alfuzosin binds to α1-adrenergic receptors with equal affinity. The reason for its prostate selectivity is not well understood. It seems to be related to the ability of alfuzosin to accumulate selectively in prostate tissue. Because of this prostate affinity, therapeutic doses of alfuzosin have little effect on systemic arterial blood pressure and are much less likely to cause syncope. IFIS remains a concern, however, with the use of alfuzosin. The drug is well absorbed and is available in a once-daily dosage form.
Tamsulosin
Tamsulosin (see Figure 7-1) is the first clinically available antagonist that blocks specific subtypes of the α1-adrenergic receptor: the α1A and α1D subtypes. The α1A-adrenergic receptor has been shown to mediate the contraction of human prostatic smooth muscle.26 Because tamsulosin has a high affinity for the α1A-adrenergic receptor, it is effectively used to treat benign prostatic hyperplasia. The selectivity of this compound for the prostate is reflected in the fact that there is little decrease in blood pressure after therapeutic doses of the drug. Tamsulosin is well absorbed after oral administration and circulates tightly bound to plasma proteins. It is extensively metabolized in the liver and excreted as inactive conjugation products in the urine. Tamsulosin is less likely to cause orthostatic hypotension and syncope than other α1-selective antagonists.7,18 Similar to the other α1-adrenergic receptor antagonists, tamsulosin use has been associated with an increased incidence of IFIS. Other adverse effects include nasal stuffiness and skin rash.
NONSELECTIVE α-ADRENERGIC RECEPTOR ANTAGONISTS
Imidazolines
Analogues of the imidazoline adrenergic amines were among the first synthetic adrenergic blocking agents to be identified. Phentolamine (Figure 7-2) is the only compound from this class that is still clinically available. It is a competitive antagonist at α1-adrenergic and α2-adrenergic receptors. It also evokes histamine release, acts as a cholinomimetic, and blocks 5-hydroxytryptamine (serotonin, 5-HT) receptors. Therapeutically, doses sufficient to achieve adrenergic blockade can produce side effects attributable to these other actions. Nevertheless, adverse responses linked to significant decreases in peripheral vascular resistance—such as hypotension—are the major problems associated with phentolamine use. Reflex tachycardia is a special concern with phentolamine. By blocking prejunctional α2 receptors, the drug interferes with the negative feedback mechanism (autoregulation) that normally limits the amount of norepinephrine released in response to an acute decrease in blood pressure. This lack of autoregulation leads to excessive transmitter release by sympathetic nerves supplying the heart, which may produce increases in heart rate sufficient to cause myocardial ischemia and cardiac arrest.27,29
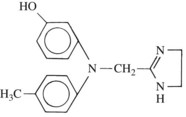
Most recently, phentolamine mesylate has been approved for the reversal of soft tissue numbness after administration of local anesthetics with vasoconstrictors for nonsurgical dental procedures. The drug is formulated in dental cartridges (0.4 mg/1.7-mL cartridge) and is injected in the same manner as the local anesthetic when pain relief is no longer needed. The median duration of post-treatment anesthesia in the upper and lower lips of adults16 and children31 is reduced by 85 minutes when phentolamine mesylate is injected at the end of restorative and dental hygiene procedures lasting about 45 minutes. Return of normal function (e.g., speaking, smiling, drinking) occurs in concert with return of normal sensation. It is unlikely that the phentolamine is acting by reversing the vasoconstrictor effect of injected epinephrine or levonordefrin, which should have already disappeared from the local tissues. Instead, the phentolamine probably increases local blood flow by blocking sympathetic tone, which hastens the removal of the local anesthetic from local neurons. Doses of phentolamine used for this purpose (0.2 mg to 0.8 mg) are approximately 10 times less than doses injected intravenously for treatment of hypertensive emergencies, and adverse effects have been similar to those reported after sham injection.
β-Haloalkylamines
Phenoxybenzamine is the only currently used member of the β-haloalkylamines. It is classified as an antagonist at α1-adrenergic and α2-adrenergic receptors. Phenoxybenzamine initially binds reversibly to the receptors, but then it undergoes a chemical reaction that allows the drug to become covalently linked. The initial binding is governed by the same chemical binding forces described in Chapter 1. During development of the blockade, the presence of an agonist, or even a competitive α-blocking drug, decreases the blocking activity of phenoxybenzamine by competing for the α-adrenergic receptors. When the block has developed completely, usually in approximately 1 hour, no drug can successfully compete for the receptor because phenoxybenzamine will have formed a stable covalent bond with the receptor. This stage of the block is referred to as irreversible or nonequilibrium and has a half-life of about 24 hours, with effects persisting for several days. Similar to phentolamine, phenoxybenzamine is nonselective and blocks the prejunctional α2 receptors responsible for regulating the release of norepinephrine. Its adverse effects are largely predicated on its long-lasting, insurmountable blockade of α receptors. In high doses, phenoxybenzamine also inhibits responses to histamine, acetylcholine, and 5-HT, however, and blocks transporter systems responsible for the tissue uptake of norepinephrine.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


