CHAPTER 6 Clinical Genetics for the Dental Practitioner
The purpose of this chapter is twofold: to review genetic principles and to mention a few examples of the influence of genetic factors on major craniofacial, oral, and dental conditions. As the basis for relatively rare developmental dysplasias, diseases, and syndromes that show a genetic cause or marked genetic influence becomes known, increasing attention is being paid to those genetic factors that influence (or are associated with) more common conditions. An increased appreciation of how genetic factors interact with environmental (nongenetic) factors to influence growth and pathology will lead to an increased understanding of pathogenesis and the recognition that some groups or individuals may be more susceptible, or that they may respond differently to treatment.1 Further information may be found online in the Genetics Home Reference Your Guide to Understanding Genetic Conditions at http://ghr.nlm.nih.gov.
REVIEW OF GENETIC PRINCIPLES
Remarkable advances in the biochemical techniques that are used to study cell molecular biology and DNA have taken researchers to the threshold of understanding the regulation of cell functions. To illustrate, not so long ago DNA analyses were performed on minute amounts (picograms) of DNA. This limitation was necessary because there was so little DNA available for study in samples. When the DNA polymerase enzyme was discovered that could replicate DNA through the polymerase chain reaction and make it by the gram, this sample problem disappeared. This advance facilitated completion of the human genome project, which resulted not only in definition of a single human genome sequence composed of overlapping parts from many humans, but also in an expanding catalogue of more than 1 million sites of variation in the human genome sequence. These variations (or polymorphisms) may be used as markers to perform genetic analysis (including analysis of genetic-environmental interaction) in human beings.2 The genome varies from one individual to the next, most often in terms of single base changes of the DNA, called single nucleotide polymorphisms (SNPs, pronounced “snips”). The main use of this human SNP map will be to determine the contributions of genes to diseases (or nondisease phenotypes) that have complex, multifactorial bases.3
CELL DIFFERENTIATION AND DEVELOPMENTAL BIOLOGY
It is fascinating that a single fertilized ovum contains within itself the potential for development of the incredibly complicated human organism. Cellular differentiation is a critical component of this developmental process, and aside from the development of antibody diversity, typically occurs in the absence of genetic alteration or mutation. Different types of cells gain their specific identities by using a particular subset of the approximately 30,000 or more genes present within the genome. The types of polypeptides that a cell can synthesize include enzymes, which catalyze various activities of cellular metabolism and homeostasis; structural proteins, which form the intracellular and extracellular scaffolding or cellular matrix; and regulatory proteins, which convey signals from the outside of the cell to the nucleus and modulate or control specific gene expression. In a developing embryo, cells reside in a three-dimensional environment and are responsive to signals from themselves (autocrine), from nearby sources (paracrine), and from anatomically distant sources (endocrine). Many of these signals are mediated by soluble molecules (either peptide or nonpeptide in origin) that bind to specific receptors (proteins) that are present on the surface or on the inside of cells. In addition to signals from soluble factors, cells can respond to cell-to-cell or cell-to-extracellular matrix signals.4
The action of “turning on” or “turning off” specific genes, referred to as regulation of gene expression, is carefully orchestrated and remains a critical element in determining cell specificity and tissue morphogenesis. Transcription factors bind to DNA and either facilitate or suppress initiation of gene transcription, the most common control point of gene expression. In the development of the craniofacial complex there is increasing evidence for the role of homeobox-containing gene families that encode transcription factors. These then are critical for the control of complex interactions between genes that are subsequently expressed during development.5
CHROMOSOMES
One area of special interest to the clinician is cytogenetics, the study of chromosomes. This interest has been stimulated by the development of techniques in which cells are grown in culture and the chromosomes are examined under a microscope for changes in size, shape, and fine structure. This is called karyotyping. Fig. 6-1 shows the karyotypes of a normal human male and female. By applying this technique, Lejeune and colleagues demonstrated that the fundamental cause in Down syndrome is the presence of an extra specific chromosome (number 21) in the affected individual’s karyotype.6 When an entire extra chromosome is present, the condition is called a trisomy of the chromosome in question, for example, trisomy 21 for Down syndrome. Fig. 6-2 shows the karyotype of a male who has Down syndrome. The extra chromosome in the group of number 21 and number 22 chromosomes is readily apparent.
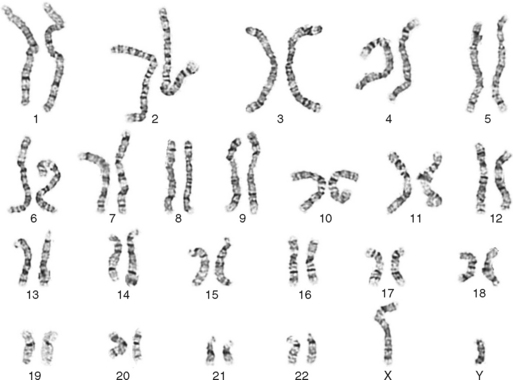
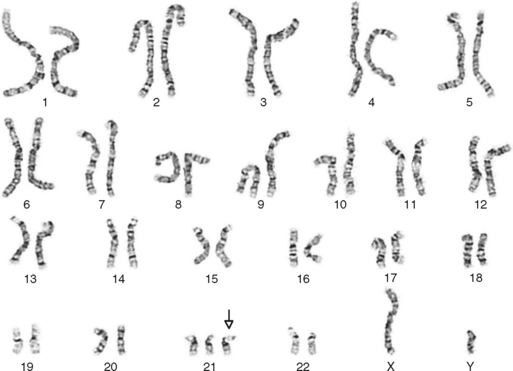
Figure 6-2 Banded karyotype of a male with trisomy of chromosome 21 (Down syndrome).
(Courtesy Cytogenetic Laboratories, Indiana University School of Medicine.)
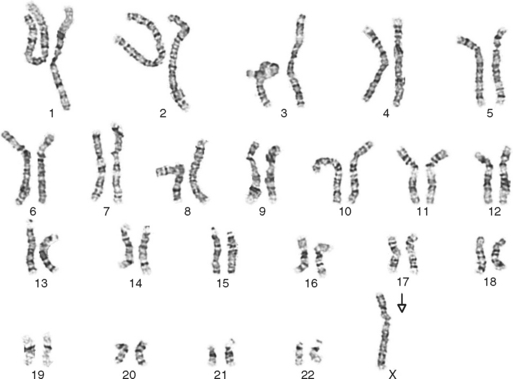
Since this report in 1959, many disease states have been shown to be associated with an incorrect chromosome complement. By using this approach with considerable refinement, it was shown that alterations in the fine structure of chromosomes, as well as in their number, could be present. Monosomy of an autosome, or a missing autosomal chromosome, had not been believed to be compatible with life, but several monosomies in live-born children have now been reported. Monosomy of the sex chromosomes can be compatible with life and typically affects development of both internal and external sex organs of the individuals. The best known example of this is Turner syndrome, which occurs in approximately 1 in every 5000 live female births. These persons are phenotypic females who are usually missing one of the X chromosomes and are chromosomally designated as 45, X. Other aberrations of the X chromosome may also cause Turner syndrome. Affected individuals are typically short of stature, lack secondary sex characteristics, and are sterile. The Turner syndrome karyotype is shown in Fig. 6-3. Table 6-1 lists common chromosomal aberrations that produce clinical disease, including examples of translocations (the attachment of a broken piece from one chromosome to another, but not homologous, chromosome) and deletions (the absence of a piece of chromosome).
| Type | Specific Alteration | Clinical Result |
|---|---|---|
| Aneuploidy | Trisomy 21 | Down syndrome |
| Trisomy 18 | Edwards syndrome | |
| Trisomy 13 | Patau syndrome | |
| Extra X chromosomes | In females: XXX, XXXX, XXXXX syndromes | |
| In males: Klinefelter syndrome—XXY, XXXY, and XXXXY | ||
| Monosomy, autosomal | Usually nonviable | |
| Monosomy, X chromosome | In females: Turner syndrome, 45,X | |
| In males: nonviable, 45,Y | ||
| Translocation | 14/21, 21/21 or 21/22 | Translocation carrier (normal phenotype) or Down syndrome |
| Deletion | Ring chromosome | Variable |
| Short arm chromosome No. 5 | Cri du chat syndrome | |
| Philadelphia chromosome (No. 22) | Chronic myeloid leukemia |
Chromosome abnormalities are an important cause of spontaneous abortion. About 15% of all recognized pregnancies end in spontaneous abortion, and the incidence of chromosome abnormalities in such abortions is greater than 50%. Only 0.3% to 0.5% of all live-born infants have a chromosome abnormality that is detectable with standard microscopic karyotyping. Microdeletions and microduplications of DNA, not visible by routine chromosome karyotype analysis, are a major cause of human malformation and mental retardation. A complementary analysis called comparative genomic hybridization (CGH) or array comparative genomic hybridization (arrayCGH or aCGH) can improve the diagnostic detection rate of these small chromosomal abnormalities. This technique attains such a high-resolution screening by hybridizing differentially labeled test and reference (“normal”) DNAs to arrays consisting of thousands of genomic clones. In this way, relatively small differences between the test and reference DNA sequences may be discovered and investigated further if indicated.7,8
HEREDITARY TRAITS IN FAMILIES
Heritability is the proportion of the total phenotypic variance in a sample that is contributed by genetic variance.9 On an individual basis for a binary trait (i.e., a disease or trait that an individual either has or does not have), heritability is not the proportion of disease or the trait attributable to, or caused by, genetic factors. For a quantitative trait, heritability is not a measure of the proportion of an individual’s score attributable to genetic factors.10 A trait with a heritability of 1 is said to be expressed without any environmental influence, whereas a trait with a heritability of 0.5 has half its variability (from individual to individual) influenced by environmental factors and half by genotypic factors. Values greater than 1 may occur because the methodology provides an estimate of heritability under several simplifying assumptions that may be incorrect.
There is the common perception that knowing a trait’s heritability will somehow affect how a patient should be treated (e.g., for malocclusion) or that it will define the limits of tooth movement or the manipulation of jaw growth. This is not true. The ability of the patient to respond to changes in the environment (including treatment), which has nothing to do with heritability, defines these limits. Heritability estimates imply nothing about trait size or treatment limits based upon a presumed genetic “predetermination.”11 Even so, the estimation of heritability can provide an indication of the relative importance of genetic factors on a trait in a group at that time. Confirming that there is a certain degree of genetic influence on a trait is a preliminary step to performing further specific genetic linkage studies (using DNA markers) to determine areas of the genome that appear to be associated with the characteristics of a given trait.12
When hereditary traits in families are to be studied, it is convenient to think of three classes of genetically influenced traits:.(1) monogenic, (2) polygenic, and (3) multifactorial. Recently the polygenic and multifactorial classes have often been combined into what are referred to as complex traits rather than Mendelian traits.13 Monogenic traits are produced and regulated by a single gene locus. Usually they are relatively rare in the general population (occurrence in fewer than 1 per 1000 individuals). However, if the appearance of an affected person is striking, there may be instant recognition of the disease, as with patients having albinism, achondroplasia, or neurofibromatosis. Monogenetic conditions often occur in families and show transmission characteristics of the Mendelian (dominant or recessive) traits.
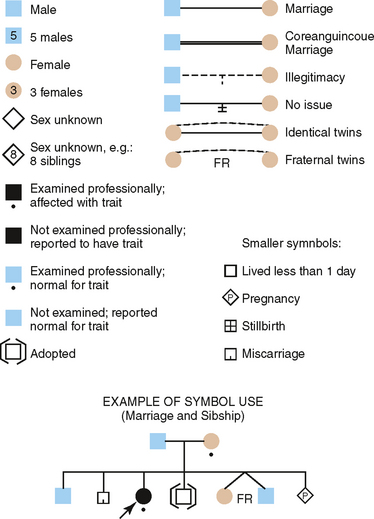
Construction of a pedigree, which is a shorthand method of classifying the family data, conveniently summarizes the family data for the study of inherited traits. The symbols used in constructing a pedigree are shown in Fig. 6-4. The observable inheritance patterns followed by such monogenic traits within families are determined by (1) whether the trait is dominant or recessive, (2) whether the gene is autosomal (on one of the autosomes) or X linked (on the X chromosome), and (3) the chance distribution in the offspring of those genes passed from parents in their gametes (sperm and ova). Pedigree construction is a valuable tool for the clinician who is concerned with the diagnosis of and counseling regarding hereditary traits. Every dentist should be able to construct and interpret a pedigree, because it is a certainty that patients will come to the dentist’s office with heritable oral diseases that need diagnosing before treatment is begun.
The simple patterns of monogenic inheritance seen in families are described in the following discussion. Because all the Mendelian modes of inheritance are found in the amelogenesis imperfecta (AI) disorders, these are used to illustrate basic genetic principles.
DEVELOPMENTAL BIOLOGY OF ENAMEL
For a review of how molecular biologists are studying the genetic factors involved in dental development, there is a paper by Tucker and Sharpe.14 The two developmentally different cell layers involved in dentinogenesis, inner enamel epithelium (enamel) and neural crest (dentin), are separated by an extracellular matrix.15 Specific tooth development is then mutually dependent on reciprocal cell-to-cell signaling between these two developmentally different cell layers.16 The genes involved in the development of these tissues are candidates for DNA mutation analysis, especially if they are in a chromosome location that has been associated with or linked to an inherited defect of enamel or dentin. The most intriguing dental research today is (1) the attempt to localize the genes for these proteins to specific loci and (2) the biochemical identification of a specific defect in the protein that prevents it from functioning normally. The following is a discussion of genetic principles best exemplified by the heritable disorders of enamel. Further discussion of the molecular basis of the heritable disorders of dentin and enamel appears in Chapter 7.
Based on the clinical appearance, radiographic characteristics, and microscopic features, oral pathologists have recognized three major types of inherited enamel defects: hypoplasia, hypocalcification, and hypomaturation.17 These terms also provide the general description of the disease phenotypes. For example, in type 1, enamel hypoplasia, the enamel is hard and well calcified but defective in amount, so the teeth appear small. Two types of deficient enamel phenotypes are seen: generalized (all the enamel) and localized (pits and grooves in specific areas). Type 2, hypocalcification disorders, are those in which the enamel matrix is so drastically altered that normal calcification cannot occur, with the result that the clinical phenotype is a soft, mushy enamel that easily wears away. Type 3 defect, hypomaturation, involves the process of maturation of the enamel crystal. This occurs after an essentially normal enamel matrix has been established. The enamel is of normal thickness (not hypoplastic) and relatively normal hardness (slightly hypocalcified) with reduced radiographic density and discoloration.
One characteristic of inherited dental defects is that both dentitions (primary and permanent) are affected. Occasionally, the defect is expressed differently in the two dentitions, as in the case of dentin dysplasia type II.18 However, it is much more common to see the same clinical and radiographic picture in both dentitions. Both dentitions are affected in the AI disorders.
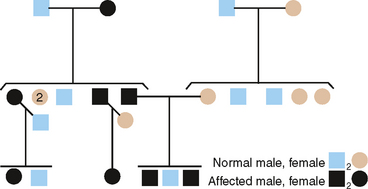
AUTOSOMAL DOMINANT INHERITANCE
From pedigrees such as that shown in Fig. 6-5, the following criteria for AD inheritance may be deduced:
AUTOSOMAL RECESSIVE INHERITANCE
Recessively inherited traits require that both genes of a given pair at a single locus code for defective proteins. Thus, of the two alleles at this genetic locus for AI, both must be mutants to show the trait. The following three gene pairs are recognized: AA—normal; Aa—heterozygote, showing an unaffected phenotype; aa—homozygous-affected. The most common genetic situation producing an affected child is that in which both parents are heterozygous at this genetic locus (Fig. 6-6).
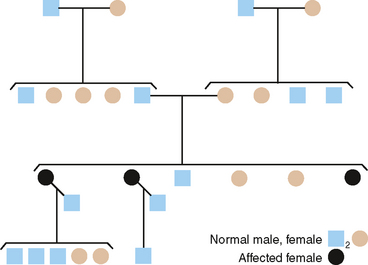
The following significant points about recessive inheritance must be noted:
Of several AR types of AI, the one chosen for discussion here is the pigmented hypomaturation form. In this instance, the genetic defect probably lies in the protein needed in late tooth development to produce mature, hard, and dense enamel. The defective enamel present is softer than normal but not nearly as soft and easily abraded as in the hypocalcification defect. Remarkably, a brown pigment is found in these outer layers of enamel that are formed last, imparting a dark brown, unsightly appearance that necessitates restorative treatment. A pedigree illustrating AR inheritance of this hypomaturation defect is shown in Fig. 6-6.
X-LINKED OR SEX-LINKED INHERITANCE
To this point we have considered heritable defects in two of the three major types of enamel disorders. The third type-AI, hypoplastic type-shows both autosomal and X-linked modes of inheritance, but only one X-linked type is described here.
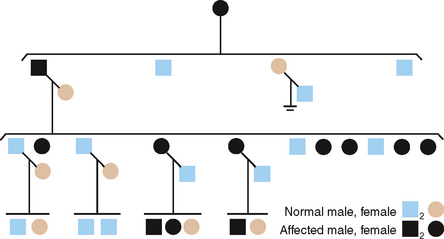
X-LINKED DOMINANT
Fig. 6-7 is the pedigree of a family with an X-linked dominant (XLD) form of AI, hypoplastic type.19 The clinical features are diagnostic and in some females can be quite striking.
X-LINKED RECESSIVE
A pedigree of a family with the X-linked recessive form of enamel hypomaturation is shown in Fig. 6-8. The genetic criteria for diagnosing an XLR trait are summarized as follows:
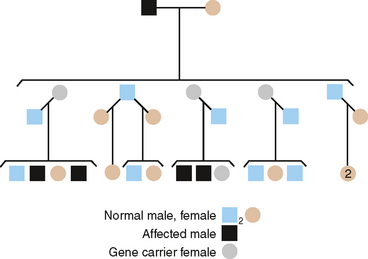
It should be noted that heterozygous females occasionally show significant clinical expression of a single XLR gene. The reason for this apparent contradiction is the process of X-inactivation, termed lyonization after geneticist Mary Lyon. This occurs only in females. All normal female cells have two X chromosomes, but most of the genes on one of the two X chromosomes are inactivated approximately at the blastula stage of development. This has the effect of making the total number of active, X-linked genes about the same in both males and females. If the female is heterozygous for an X-linked trait, two populations of cells result. One cell population has genes on one X chromosome that are active, while the other cell population has genes on the other X chromosome that are active. When by chance the X chromosome with the deleterious gene is active in a significant proportion of the cells, its expression may be observed in that female. Chance dictates that this imbalance does not occur frequently, but because all females are, by definition of lyonization, mosaic with regard to X-linked traits, phenotypic expression of heterozygous genes may occur in them.
The previous statements concerning the distribution of XLR genes in males and females apply equally as well to XLD genes. The principal difference lies in the fact that when the gene is dominant more females than males will show the trait (see pedigree in Fig. 6-8). Because all XLR genes behave as dominant genes in males, no new criteria are made for their inheritance in males. The following criteria distinguish an XLD trait in families:
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


