CHAPTER 6 Adrenergic Agonists
HISTORY
The first recorded study of an adrenergic agent resulted in the isolation in 1887 of ephedrine from the herb ma huang, which had been grown and used in China for centuries. At the same time, investigators were making extracts of all the organs of the body in an attempt to discover new hormones. Studies by Oliver and Schafer in the early 1890s showed a potent vasopressor substance in extracts of the adrenal gland. The active agent, epinephrine, was soon isolated by Abel, prepared commercially, and marketed in the United States under the trade name of Adrenalin(e). By 1905, it had been synthesized and was being incorporated with local anesthetics. In that year, an account was published of the results of mixing procaine with epinephrine to obtain dental anesthesia.3 1887 also witnessed the synthesis of amphetamine, which was first marketed in the 1930s and became widely abused by the mid 1950s. The addictive nature of amphetamine was soon realized and led to its designation in 1970 as a drug of high abuse potential. Concerns in more recent years regarding the potential adverse cardiovascular effects of various sympathomimetic drugs have prompted the U.S. Food and Drug Administration to regulate increasingly their use as appetite suppressants, nasal decongestants, and cold remedies. In 2004, ma huang was banned for sale as a dietary supplement.
CLASSIFICATION OF ADRENERGIC DRUGS AND RECEPTORS
Adrenergic receptors have been classified into three major types: α1-adrenergic, α2-adrenergic, and β-adrenergic receptors. In recent years, numerous receptor subtypes (α1A, α1B, α1D; α2A, α2B, α2C; β1, β2, β3) have been discovered by molecular cloning and pharmacologic techniques.4,20,36,43 Several dopamine receptors have also been identified (D1, D2, D3, D4, D5).15,45 These receptor subtypes are distinguished by differences in their amino acid sequences, as determined from gene-cloning experiments,4,45 and by their affinity for subtype-selective drugs. Many adrenergic agonists activate more than one of the major adrenergic receptor types. In contrast, some agonists selectively activate α receptors, others activate β receptors, and some are selective for an individual adrenergic receptor subtype (e.g., β1 or β2). Similarly, as discussed in Chapter 7, there are antagonists for the various adrenergic receptors, some of which are receptor type or subtype selective, and some of which are nonselective. The development of receptor-selective agonists and antagonists remains an active area of research.33,37,40,43
CHEMISTRY AND STRUCTURE-ACTIVITY RELATIONSHIPS
The chemical structures of the three endogenous adrenergic amines—dopamine, norepinephrine, and epinephrine—are illustrated in Figure 6-1. These compounds are synthesized sequentially in adrenergic nerve terminals and adrenal chromaffin cells (see Chapter 5). These three agents, all derived from tyrosine, are also referred to as catecholamines because they are catechol derivatives of phenylethylamine.
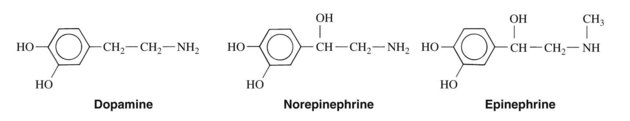
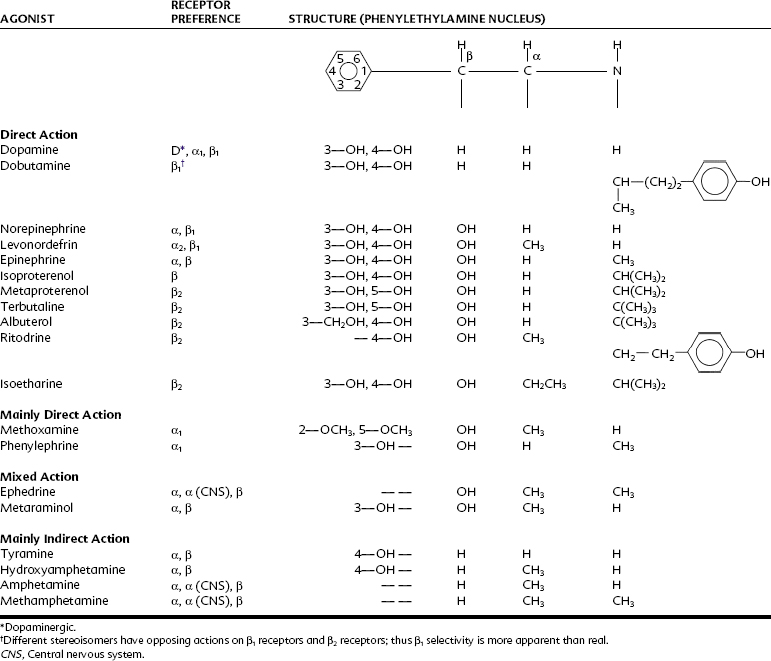
Table 6-1 lists some adrenergic agonists currently in use and illustrates certain major alterations in biologic activity that occur with structural modifications. The following conclusions about the relationship between structure and activity can be drawn:
PHARMACOLOGIC EFFECTS
The pharmacology of the adrenergic agonists is complicated by the diversity of the drugs in this group. They differ in mode of action (direct, indirect, or mixed), receptor selectivity, and relative predominance of peripheral and CNS effects. Predicting the pharmacologic activity of any adrenergic agonist is possible by knowing whether it is direct acting or indirect acting, and what receptors it affects. The density of the receptor population in a particular organ or organ system also influences the effectiveness of adrenergic agonists. A smooth muscle with a high density of α-adrenergic receptors would be strongly contracted by a drug that is efficacious in activating α-adrenergic receptors, but another smooth muscle, expressing few or no α receptors, would be minimally or not at all affected by the same agonist. Table 6-2 summarizes the relative receptor preferences of several adrenergic drugs.

Endogenous Catecholamines: Norepinephrine and Epinephrine
Vascular effects
Figure 6-2 shows the typical cardiovascular responses to the intravenous bolus injection of these catecholamines. The qualitatively different effects of high versus low doses of epinephrine on blood pressure and heart rate described earlier are apparent as the initially high concentration of drug declines over the course of several minutes into the low dose range.
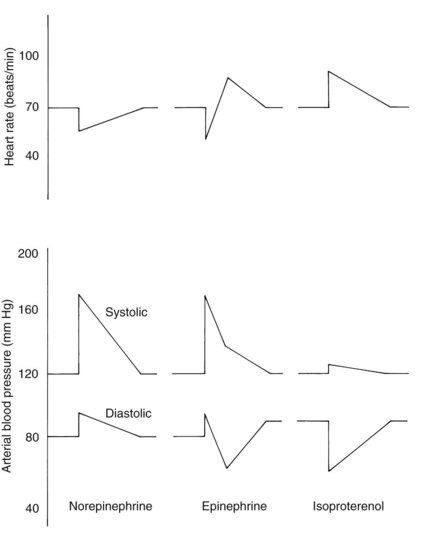
Cardiac effects
All the effects described are effectively antagonized by β receptor blockade. Simulation of α1-adrenergic receptors has been shown to enhance myocardial contraction and to prolong the refractory period, however, and has been implicated in certain ventricular arrhythmias occurring during general anesthesia.41
Stimulation of β-adrenergic receptors increases the work of the heart, which elevates cardiac oxygen consumption. Overall, cardiac efficiency (cardiac work done relative to oxygen consumption) is diminished. The delivery of oxygen to the heart by the coronary arteries is variably affected by the relative amounts of α and β receptor activation produced by each adrenergic agonist (see Table 6-2) and by metabolic regulators of local blood flow.
Metabolic responses
Metabolic responses to β2-adrenergic and β1-adrenergic receptor stimulation lead to a transitory increase in circulating blood glucose as a result of liver glycogenolysis and increased glucagon secretion.1 An α2 receptor–mediated inhibition of insulin secretion contributes to the hyperglycemia caused by epinephrine. Stimulation of β1 and β3 receptors is involved in the hydrolysis of triglycerides, causing an increase in triglyceride lipase activity and subsequently in the concentration of circulating free fatty acids. The specific receptors that mediate metabolic effects vary among species.
Dopamine
Cardiovascular effects
Dopamine interacts with various receptor types to influence vascular function, and is used therapeutically for maintaining renal function in cases of shock associated with compromised cardiac output. Although “low-dose” dopamine has now been used in this manner for more than 30 years, there is currently a growing body of evidence suggesting that this use for dopamine should be abandoned. Early data using indirect clearance measurements of blood flow suggested that dopamine stimulates vascular D1 receptors to dilate selectively the renal, celiac, hepatic, and mesenteric vasculatures, and that increases in glomerular filtration rate and Na+ excretion occur in conjunction with increased renal blood flow.16 With moderate doses, dopamine was thought to act at myocardial β1-adrenergic receptors to increase contractile force. Critics of low-dose dopamine have suggested that this increase in cardiac index, rather than dilation of the renal vasculature, is the primary reason for any observed increases in renal blood flow with low-dose dopamine,44 and that the potential for harm outweighs the benefits.30 At higher doses, dopamine also stimulates α1-adrenergic receptors, which produces vasoconstriction. As with all catecholamines, excessive doses of dopamine can cause tachycardia and generate arrhythmias. In addition to stimulating α1 and β1 receptors directly, dopamine in moderate to high doses causes the release of norepinephrine from sympathetic nerve terminals.
Fenoldopam, a pharmacologic congener of dopamine, selectively activates D1 receptors at therapeutic doses. It decreases mean blood pressure, increases renal blood flow, and causes diuresis and natriuresis. It is used intravenously for acute treatment of severe hypertension (see Chapter 28).
Other effects
Dopamine is involved with the sensory division of the autonomic nervous system. The high concentration of dopamine in the glomus cells of the carotid body and the effects of hypoxia on these cells suggest that dopamine is an inhibitory transmitter that modulates the frequency of discharge of the sensory fibers from that structure.19 It is theorized that, by this mechanism, dopamine may affect cardiovascular and respiratory responses.
Dopamine itself does not penetrate the blood-brain barrier. Levodopa, which is converted into dopamine, does enter the CNS, however, and is used to treat Parkinson’s disease (see Chapter 15). Approximately 95% of an oral dose of levodopa is normally decarboxylated in the periphery to dopamine,8 leading to significant peripheral side effects attributable to dopamine. Dopamine can also produce nausea and vomiting as a result of excitation of the medullary chemoreceptor trigger zone, which lies outside the blood-brain barrier.
α-Adrenergic Receptor Agonists
The group of drugs classified as α-adrenergic receptor agonists is growing increasingly diverse. These drugs stimulate α-adrenergic receptors, but have low affinity for β-adrenergic receptors. Phenylephrine and methoxamine differ from epinephrine and norepinephrine by being selective agonists at α1-adrenergic receptors. Their primary pharmacologic effect is to cause contraction of vascular smooth muscle, resulting in an increase in systolic and diastolic blood pressures and reflex bradycardia. They are often administered either intranasally or systemically for temporary relief from nasal congestion. Because these drugs increase blood pressure, safety is always a concern. The α-adrenergic receptor agonist phenylpropanolamine was widely used in over-the-counter cold remedies until research studies showed that it increased the risk of hemorrhagic stroke in women,25 which led the U.S. Food and Drug Administration (FDA) to mandate its removal from these medications. Other agonists with actions similar to phenylephrine and methoxamine include metaraminol, although it is a mixed-acting agonist (discussed later) because it releases catecholamines in addition to directly stimulating α1-adrenergic receptors. Midodrine is a newer synthetic drug that selectively activates α1-adrenergic receptors. It also causes vasoconstriction, and is used to treat postural hypotension caused by impaired autonomic nervous system function.
The α2-adrenergic receptor agonists clonidine, guanabenz, guanfacine, and methyldopa (Figure 6-3) effectively enter into the CNS and stimulate α2-adrenergic receptors in the brain. They are, in varying degrees, selective agonists at α2 receptors. Methyldopa, an α-methyl derivative of dopa (dihydroxyphenylalanine, an important intermediate in the synthesis of norepinephrine), enters into the nerve terminal and is converted into the α2 receptor–selective agonist α-methylnorepinephrine by the same synthetic process that converts dopa into norepinephrine. Although α-methylnorepinephrine is present in neuronal storage vesicles in peripheral sympathetic nerves, this metabolite of methyldopa is nearly equipotent to norepinephrine as a vasoconstrictor in humans. This agent has been developed as the drug levonordefrin, which is used as a vasoconstrictor in local anesthetic solutions. Clonidine was first used as a nasal decongestant, but it was soon found to decrease blood pressure. An imidazoline derivative, clonidine is a selective α2-adrenergic receptor agonist with relatively weak peripheral effects. Guanabenz and guanfacine are guanidine derivatives that, similar to clonidine, also selectively activate α2-adrenergic receptors.
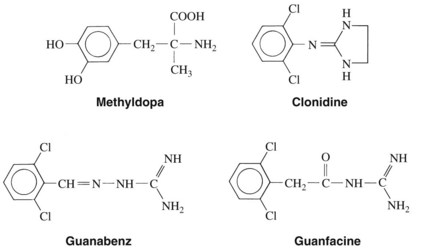
The administration of these centrally acting drugs in humans results in moderate decreases in mean arterial blood pressure. This effect usually occurs without increases in heart rate because a decrease in CNS sympathetic outflow tends to reduce venous return, heart rate, and cardiac output. Guanfacine decreases peripheral vascular resistance without affecting cardiac output.18 Intravenous administration of these drugs may increase blood pressure acutely as a result of stimulation of peripheral vasoconstrictor α2 receptors. This effect is not usually seen with oral administration.
Serendipity has played a role in the use of clonidine to treat the withdrawal symptoms of opioid addiction.29 Clonidine, when given to addicts undergoing withdrawal, blocks the nausea, vomiting, sweating, diarrhea, and other symptoms of excessive autonomic discharge (see Chapter 51). Evidence indicates that either systemic or intracerebral injection of opioids inhibits neuronal activity in the locus ceruleus of the dorsolateral pons. When the opioids are withdrawn, certain neurons are thought to be disinhibited and to release excessive norepinephrine, which gives rise to the symptoms of withdrawal. Clonidine, by stimulating presynaptic α2 receptors on these same neurons, />
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


