CHAPTER 52 Toxicology
GENERAL PRINCIPLES
All chemical substances can cause harm or kill if encountered at sufficiently large concentrations over crucial periods of time. This statement embodies insight articulated by Paracelsus in the sixteenth century that dose is the major determinant of toxicity. A subset of substances has relatively specific toxic effects, however. These are considered very harmful based on human experience and are considered poisons or toxins. Beyond this base of experience exists a vast number of uncharacterized, potential toxicants. As of 2007, the Chemical Abstracts Service10 reported counts of more than 32 million organic and inorganic substances, 245,000 inventoried or regulated substances, and some 15 million commercially available chemicals. Because many of these are potentially toxic, this array dictates that toxicologists use some means of triage toward assessment of potential toxicants. At present, selection of chemicals for toxicologic testing is dictated by their potential for use, by funding of basic research on the chemicals, and initial evidence of their specific adverse effects.
The ultimate aim of toxicologic science in society is to guide safe use of chemicals. The definitions in Box 52-1 can assist in understanding and promoting concise communication in the approach to this objective. Safety is a negative entity—that is, the absence of threat of injury. As such, safety cannot be proved directly. Society often simplistically considers chemicals “safe” or “toxic.” Such naive characterization can preclude the rational judgment that enables safe uses of chemicals. Critical judgment requires understanding of the distinction between the terms toxicity and hazard to enable assessment of risks (see Box 52-1). Toxicologic assessment promotes safety by defining hazardous situations of use so that the unsafe use of chemicals can be avoided.
| Safety | Condition of being secure from threat of danger, harm, or injury |
| Toxicity | Property of grave harmfulness or deadliness associated with a chemical that is expressed on biologic exposure |
| Hazard | Threat of danger directly related to circumstances of use of a chemical |
| Risk | Expected frequency of occurrence of an adverse effect in a given situation |
The criteria developed by Sir Austin Bradford Hill36 provide a sound basis for consideration of causal relationships and should be considered a touchstone for expert opinion regarding cause and effect (Box 52-2). None of these criteria should be considered as absolutely essential, and they cannot be considered as proof of causal relationships. Their careful application during evaluation of potential cause-and-effect relationships can assist, however, in organizing knowledge toward a weight-of-evidence judgment and may provide an alternative interpretation for consideration.
BOX 52-2 Hill Criteria for Consideration of Causal Relationships
From Hill A: The environment and disease: association or causation? Proc Roy Soc Med 58:295, 1965.
| CRITERION | EXPLANATION |
|---|---|
| Strength of association | Observed magnitude of the association compared with other relevant observations should be considered as a primary indicator in assessment of cause and effect |
| Consistency | Association of cause and effect can be observed repeatedly by others under appropriate circumstances |
| Specificity | Particular conditions produce the effect, or a specific group is affected. Bounds of causal relationship should be delimited |
| Temporality | Causation generally occurs before effect, whereas correlational effects can vary in temporality |
| Biologic gradient | Demonstration of a fundamental dose relationship provides convincing evidence of cause and effect |
| Plausibility | Some basis in previous knowledge provides a means of common understanding (remember, however, that all phenomena were novel at some point) |
| Coherence | Care should be taken that interpretation of cause and effect does not unduly conflict with scientifically established facts of biology and medicine |
| Experiment | Manipulation of accessible variables in the potential cause-and-effect relationship has an effect |
| Analogy | Previously understood examples provide basis for formulation of testable hypotheses |
Dose-Response Relationships
Figure 52-1 presents three modes of display of idealized dose-response data to illustrate and describe the dose required for median response in subjects tested. These data are typical of quantal or all-or-nothing responses such as lethality. In this example, the dose axis is logarithmically spaced, and the data describe a log-normal distribution. Responses that arise from mass action, such as reversible occupancy of receptor by drugs, often are most easily plotted on a logarithmic axis. Alternatively, effects caused by limited biologic capacity, such as irreversible enzyme inhibition, can exhibit abrupt threshold-like effects and may be more easily analyzed on a linear dose axis. The rule is to plot the data to see what type axis is most applicable.
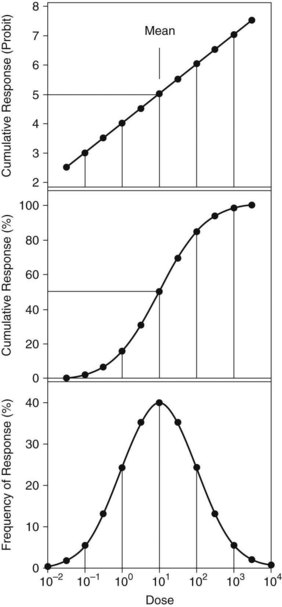
The lower panel of Figure 52-1 indicates distribution of responses across the dose axis, with a mean of 10 and standard deviation (SD) of one log10 unit. Response percentages include approximately 68.3% within ±1 SD of the mean, 95.5% within ±2 SD, and 99.7% within ±3 SD of the mean. The distribution indicates hypersusceptibility for individuals at the lowest doses and resistant responders at the highest doses. Such a plot gives a convenient way to visualize the distribution of responses across dose within the test groups.
The middle panel of Figure 52-1 plots the cumulative response versus dose across all treated groups. Here the response data are practically linear in the range from −1 SD to +1 SD for these ideal data. This plot provides convenient, accurate estimation of dose required for a 50% response, such as the median lethal dose (LD50). Real data are rarely so well behaved, however, because too few animals may be included for adequate definition of the sigmoid curve. Another disadvantage is that the sigmoid curve presents difficulty in estimating doses that elicit extremes of response, such as 1% or 99%.
An alternative presented in the top panel of Figure 52-1 uses the probit transform45 for the cumulative response. Probit units are derived by conversion of cumulative response percentages to units of deviation from the mean. The scale uses normal equivalent deviation units (NED), for which the mean is arbitrarily set to a NED value of 5 to give positive values along the axis. As is evident in the example, the probit transform linearizes the extreme values of the response function, which allows accurate estimation of doses affecting 1% or 99% of subjects exposed. In addition, the probit transform facilitates determination of the slope, which enables comparison of the dose-response function with other agents or responses.
Such plots are inadequate in dealing with issues of societal risk, however, for which policy often requires estimation of exposure posing a theoretic risk of 1 in 1 million, otherwise described as a 10−6 risk factor. Practical problems intervene, including the impracticality of experimental studies involving sufficient animals to define adequately the dose-response function at low response levels. A classic toxicologic experiment conducted at the National Center for Toxicological Research illustrates this point. Officially termed the ED01 Study,33 this experiment examined in detail the response function of mice treated with low doses of the experimental carcinogen 2-acetylaminofluorene. The study, sometimes termed the megamouse study, involved more than 24,000 mice to determine, with precision, the dose effective in producing a 1% tumor rate. This work advanced toxicologic understanding of the complexity of genotoxic and proliferative cellular events in chronic cancer bioassays. It also exhibited logistic difficulties in conducting statistical studies of low incidence and illustrated gaps in the evolving understanding of chemically induced cancer.
Factors That Change Dose-Response Relationships
Dose-response relationships can vary with many factors, including differences within and among individuals. As described in Chapter 3, factors responsible for dose-response variations within an individual over time may include age and nutritional status, environmental influences, functional status of organs of excretion, concomitant disease, and various combinations of factors. Changes in pharmacokinetics of toxicants are a frequent basis for altered dose-response relationships. Known influences include increased toxicant bioactivation by enzyme induction,31 such as occurs in certain variants of cytochrome P45013 with exposure to phenobarbital or polychlorinated biphenyls. Conversely, inhibition of metabolic clearance is possible with interacting chemicals, increasing the pharmacodynamic action of drugs and chemicals.
Cytochrome P450 variant 3A4 is an important enzyme in human drug metabolism, and its presence in the gut and liver subjects it to inhibition by many drugs and dietary components, such as grapefruit juice.19 Conversely, substances are often less toxic by the oral route when administered with food as a consequence of less rapid absorption. The time and frequency of administration can be important in altering dose-response relationships through functional changes. Many compounds induce tolerance on repeated administration, whereas others can become more toxic with closely repeated administration. Receptor densities and sensitivity may vary with time or as a consequence of previous exposure. An example of the latter is the well-known tolerance that develops to long-term administration of opioids.
Responses among individuals differ as a consequence of different genetic traits, a subject of intense interest as knowledge emerges from the Human Genome Project, and use of efficient molecular techniques and transgenic animals becomes widespread in research. Recognition and understanding of relevant aspects of human diversity derived from functional genomic research offer potential for therapeutic gains.26 The rationale is to use appropriate drugs in patients best suited to benefit, and to reduce use in patients with genetic traits that might result in toxicity. These efforts have spawned new terms, including pharmacogenetics, representing characterized genetic differences in drug metabolism and disposition, and pharmacogenomics, used to describe the broad spectrum of genes that affect drug response. A summary is available25 that describes progress in determining genetic polymorphisms relevant to drug action and disposition. Known variants linked to altered drug effects in humans include phase I cytochrome P450 enzymes, phase II enzymes such as N-acetyltransferases and glutathione-S-transferases, small molecule transporters, drug and endogenous substrate receptors, and ion channel variants. Chapters 2 and 4 provide additional information on these topics.
Similar advances are likely to be applied to understand genetic differences that result in toxic effects aside from those that arise during drug therapy. Approximately 400 million individuals worldwide exhibit a heritable deficiency in the cytoplasmic enzyme glucose-6-phosphate dehydrogenase. Because this enzyme is essential to the cell’s capacity to withstand oxidant stress through production of reducing equivalents, sensitive individuals with this enzymatic deficiency have chemically mediated hemolytic anemia when exposed to oxidants.6
Acute Versus Chronic Toxicity
Chronic toxicity may exhibit little or no apparent relationship to acute toxicity. In such cases, understanding of cause and effect requires careful study. Of the many examples of chronic toxicity, carcinogenesis currently is of greatest concern in society. Precancerous cellular changes occur and develop slowly and may remain undetected over long periods. Periodic dental examinations often play a significant role in detection of cancers of the oral cavity. Knowledge of patient habits with adverse potential health effects, such as the link between tobacco use and occurrence of oral lesions,75 assists the dental practitioner in being vigilant against such chronic toxicity.
Chemically Related Toxicants
Understanding of chemical toxicity requires knowledge of related chemicals that may be present as impurities because of manufacturing or exist as a result of environmental effects. A classic example is 2,3,7,8-tetrachlorodibenzo-p-dioxin (dioxin, or TCDD), which was discovered in the herbicide mixture known as Agent Orange used in the Vietnam War. Although dioxin existed at low part-per-million levels in the herbicide mixture, the extreme toxicity of this contaminant in certain species created grave concern for contaminated areas. This concern led to a ban on the use of the herbicide 2,4,5-trichlorophenoxyacetic acid because TCDD is formed through a condensation reaction involving two molecules of 2,4,5-trichlorophenol. Dioxin also can be formed from other sources, such as combustion of municipal waste, iron ore sintering, and wood pulp and paper mills. The toxic actions of TCDD are mediated through its binding to the aryl hydrocarbon nuclear receptor,57 which regulates transcription of genes encoding cytochrome P450 enzymes in the CYP1A subfamily and several other genes that regulate cell growth and differentiation. Despite its extreme toxic potential in some species, epidemiologic studies regarding the effect of TCDD exposure on humans have been inconclusive to date.
The consequences of metabolism of drugs and chemicals after ingestion are extremely important. The following example illustrates the importance of understanding toxic effects relative to drug metabolism. Terfenadine is a nonsedating histamine H1 receptor antagonist that was widely used for relief of symptoms of seasonal allergy. This drug was removed from the market because studies revealed cardiotoxicity when terfenadine was given with erythromycin.93 The toxic interaction was traced to the antibiotic’s inhibition of the high-affinity oxidative enzyme system CYP3A in human liver and intestinal membranes.19 This interaction inhibited normal clearance of terfenadine, and the abnormally elevated concentrations produced toxicity in the form of a prolonged QT interval and the cardiac arrhythmia torsades de pointes. This antihistamine has been replaced with its active metabolite, fexofenadine, which apparently does not elicit this toxicity (see Chapter 22).
Target Organ Systems
Nervous system
A remarkable cell-selective neurotoxicant is 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), an impurity discovered42 after attempted illicit synthesis and injection of a meperidine analogue. This compound is a protoxicant for 1-methyl-4-phenylpyridinium, which is formed by monoamine oxidase and concentrated by high-affinity carrier into dopaminergic neurons. The molecular target of 1-methyl-4-phenylpyridinium is reduced nicotinamide adenine dinucleotide dehydrogenase, and the interaction blocks the cellular respiratory exchange of electrons in mitochondria of cells. Its toxic actions result in destruction of dopaminergic neurons in the substantia nigra (see Chapter 15). Death of these cells produces symptoms strikingly similar to Parkinson’s disease, leading to loss of willful motor actions.
Loss of integrity of neuronal cell metabolism can alter neuronal architecture, particularly the myelin sheath of peripheral neurons. Such effects are common to many forms of toxicity expressed in the nervous system. Various compounds, such as tri-o-cresyl phosphate, acrylamide, and metabolites of hexane, cause degeneration of long axons that control neuromuscular activities. Termed distal axonopathy,39 this toxicity involves a “dying back” or retrograde degeneration of distal axons and leads to loss of control of motor functions such as gait. Other effects, such as sensory neuropathy and paresthesia, can result from similar effects of toxicants on small sensory fibers.
Blood and hematopoietic system
Because of the crucial roles of the elements of blood in delivering oxygen and maintaining immune function, toxic effects on blood or the hematopoietic system can be life-threatening. Of these, perhaps no poisoning is more common, preventable, or treatable with timely therapy than the toxic interaction of carbon monoxide (CO) with hemoglobin (Hb). This interaction blocks the vital oxygen-carrying capacity by formation of carboxyhemoglobin (CO-Hb). Characteristics of CO and its toxic effect on various tissues sensitive to anoxia have been concisely reviewed.86 Details of treatment, which involves displacement of Hb-bound CO with oxygen, are provided in the comprehensive text Medical Toxicology.89 In mild (CO-Hb <30%) or moderate (CO-Hb 30% to 40%) cases, therapy includes use of 100% oxygen by nonrebreathing mask until CO-Hb is less than 5%. Severe poisoning can mandate hyperbaric use of oxygen to hasten the exchange.
Another toxic effect that alters the oxygen-carrying capacity of erythrocytes is the formation of methemoglobin. In this toxicity, the heme iron is oxidized from the ferrous (Fe++) to ferric (Fe+++) state by exposure to oxidizing chemicals such as nitrites or aromatic amines.92 As with CO-Hb, methemoglobin is incapable of carrying molecular oxygen to tissues. Although the effects of resultant anoxia are similar, the treatment differs. The treatment involves use of methylene blue, as a precursor to its metabolite leukomethylene blue, a cofactor that enables erythrocytes to reduce methemoglobin in the presence of reduced nicotinamide adenine dinucleotide phosphate (NADPH). This therapy has complications of potential hemolysis for treatment of infants and individuals with glucose-6-phosphate dehydrogenase deficiency92 because this enzyme is essential in the production of NADPH.
Other adverse effects on the hematopoietic system include overexpression of certain types of cells, such as that noted in the development of acute myelogenous leukemia from benzene. Benzene is a toxicant commonly encountered in petroleum distillates such as gasoline and is considered a causative agent in human leukemia, probably through an active hydroquinone38 or benzoquinone53 metabolite. The process of leukemia development seems to involve preferential selection and clonal expansion of stem and progenitor cells through interaction of the toxic benzene metabolites by multiple independent genetic and epigenetic factors.
Respiratory system
Compounds that are rapidly absorbed or highly caustic generally affect the nasal passages. Formaldehyde has a detectable pungent odor at concentrations greater than 0.5 ppm and is highly irritating to the nasal passages. The nasopharyngeal region serves as a filter for particles 10 to 30 µm in diameter. Many of these particles are cleared upward by mucociliary action. Highly water-soluble gases, such as sulfur dioxide, dissolve in moisture present in the upper respiratory membranes and form irritating sulfurous acid. Less soluble compounds, such as oxides of nitrogen and ozone, penetrate more deeply into lung and generally exert effects at membranes in the smallest airways or alveoli. Particles smaller than 5 µm may travel well down into the bronchiolar region, whereas fine particles of 1 µm nominal size reach the alveolar region.91
Chronic injury to the lung may result from inhalation of fine particles. Phagocytic mechanisms attempting to remove insoluble particles may produce tissue scarring and interstitial fibrosis, in which collagen fibers replace normal membranes and occupy alveolar interstitial space. This kind of injury is common with inhalation of silicate particles such as asbestos.64 These actions produce inflexible tissue, diminish surface area, and lead to poor surfaces for gas exchange. Another chronic lung toxicity is emphysema; its major cause is cigarette smoking. This toxic effect produces distended, enlarged air spaces that are poorly compliant but without fibrosis. The pathogenesis of this condition is not fully understood, but an imbalance between proteolytic activities of lung elastase and antiproteases seems to be involved.3 Lung cancer became a major concern with the increase in popularity of smoking; this health scourge of today was a rare disease a century ago. Smoking is believed to be the most important risk factor for this disease, presenting a 10-fold and 20-fold increase in risk for average and heavy smokers.91
Organs of excretion
As previously noted, various membrane and cytosolic enzymes in the liver provide the essential metabolic functions of oxidation and glucuronide, sulfate, and mercapturate conjugation for removal of toxicants. These reactions usually detoxify compounds, but occasionally metabolic products exhibit enhanced toxicity. Interactions can occur among effects of toxicants within the liver through induction of enzymes or depletion of metabolic resources. Acetaminophen has been widely used as an over-the-counter analgesic without adverse effects on the liver at therapeutic doses. In circumstances of glutathione depletion, however, which occurs with large acetaminophen overdose, malnutrition, or CYP2E1 induction by long-term ethanol use, a reactive electrophilic intermediate forms in sufficient amounts to produce covalent adducts that severely damage the liver (see Chapter 21).
PREVENTION AND MANAGEMENT OF POISONING
Diagnosis and treatment of poisoning are the purview of the physician. Principles of therapy for poisoning are summarized in Box 52-3 and apply to the management of any drug overdose. A dentist may be called on to provide emergency treatment of acute poisoning, however, within the practice environment or because of training as a health care professional.
Principles of Therapy for Poisoning
Terminate absorption
Any obvious means of contact with the poison should be removed. For dermal exposure to chemicals, removal of contaminated clothing and repeated washing with soap and water are indicated. With ingested compounds other than petroleum products and corrosive substances, vomiting can be induced, but only in a conscious patient. Vomiting should not be induced for poisoning by petroleum products or agents producing loss of consciousness because of the risk of aspiration. Likewise, corrosive damage to the esophagus and gastric perforations may result from corrosive substances if emesis is induced. Modern practice is to avoid inducing vomiting because it does not reliably remove ingested poisons. Gastric lavage can be used by qualified personnel if care is taken to avoid aspiration of stomach contents by the victim. Prevention of absorption of many drugs within the gastrointestinal tract can be achieved by activated charcoal (10 to 50 g in water), and cathartics may be used to hasten the exit of drugs from the intestine. The merits and limitations of gastrointestinal decontamination measures have been reviewed in detail.15
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


