CHAPTER 40 Antifungal and Antiviral Agents
ANTIFUNGAL AGENTS
Systemic fungal infections are subdivided into two groups according to the status of the patient and the type of infecting organism. Opportunistic mycoses occur in debilitated and immunocompromised patients, such as patients with acquired immunodeficiency syndrome (AIDS), leukemia, or lymphoma, and in patients who are receiving immunosuppressive agents or broad-spectrum antibiotics. Fungi involved include Candida, Aspergillus, and Cryptococcus species and various Phycomycetes. They are particularly dangerous and carry a high mortality rate.59 Endemic mycoses are caused by various pathogens distributed unevenly throughout the world and have a low incidence in temperate climates. Examples of endemic mycoses that occur in the United States include blastomycosis, histoplasmosis, coccidioidomycosis, and sporotrichosis.
A number of antifungal agents have been developed (Table 40-1). Two polyene antibiotics are amphotericin B, an important drug for many deep mycoses,1 and nystatin, an agent useful in the treatment of oral candidiasis. A third polyene, natamycin, is limited to ophthalmologic use. Miconazole, ketoconazole, and clotrimazole are representative imidazole antifungals. First introduced in 1981, ketoconazole was a major advance in systemic antifungal therapy. Clotrimazole has become a widely used topical agent. Itraconazole and fluconazole are triazole derivatives. Voriconazole and posaconazole are newer additions to the broad-spectrum triazoles that are valuable for severe fungal infections in immunocompromised patients. A new class of antifungals known as echinocandins comprises caspofungin, micafungin, and anidulafungin; these agents exhibit fungicidal activities against many fungal isolates.
TABLE 40-1 Mechanisms of Action and Clinical Uses of Some Antifungal Agents
| ANTIFUNGAL AGENT | MECHANISM OF ACTION | CLINICAL USES |
|---|---|---|
| Amphotericin B | Binding to ergosterol of fungal membrane | Topical: superficial candidiasis; intravenous: severe, progressive systemic fungal infection* |
| Nystatin | Binding to ergosterol of fungal membrane | Topical: oral candidiasis |
| Clotrimazole | Inhibition of ergosterol synthesis | Topical: oral candidiasis, superficial fungal infections† |
| Fluconazole | Inhibition of ergosterol synthesis | Oral and intravenous: systemic and localized candidiasis, cryptococcal meningitis, systemic blastomycosis, coccidioidomycosis, and histoplasmosis |
| Itraconazole | Inhibition of ergosterol synthesis | Oral: systemic fungal infections,* dermatophyte infections and sporotrichosis |
| Miconazole | Inhibition of ergosterol synthesis | Topical: cutaneous candidiasis and vulvovaginitis, superficial fungal infections† |
| Flucytosine | Inhibition of nucleic acid synthesis | Oral: systemic candidiasis and cryptococcosis |
| Griseofulvin | Disruption of mitotic spindle | Oral: dermatophyte infections of skin, hair, and nails |
| Caspofungin | Inhibition of fungal cell wall synthesis | Intravenous: severe, invasive aspergillosis, esophageal candidiasis, candidemia |
| Micafungin | Inhibition of fungal cell wall synthesis | Intravenous: prophylactic antifungal therapy in neutropenic HSCT patients, esophageal candidiasis, candidemia |
| Anidulafungin | Inhibition of fungal cell wall synthesis | Intravenous: esophageal candidiasis, candidemia |
| Terbinafine | Inhibition of ergosterol synthesis | Oral and topical: dermatophyte infections of skin, hair, and nails, and sporotrichosis |
HSCT, Hematopoietic stem cell transplantation.
* Systemic fungal infections include aspergillosis, blastomycosis, candidiasis, chromomycosis, cryptococcosis, coccidioidomycosis, histoplasmosis, paracoccidioidomycosis, phycomycosis, and sporotrichosis. Indications for specific drugs vary.
† Superficial fungal infections caused by pathogenic dermatophytes, yeasts, and Malassezia furfur.
Polyene Antifungal Drugs
Polyene antifungal drugs consist primarily of amphotericin B and nystatin, which are among the earliest antifungal drugs that became available for clinical uses. These drugs show a wide spectrum of antifungal activity against common superficial and deep fungal infections, such as candidiasis, aspergillosis, zygomycosis, and cryptococcosis.24 The primary mode of their antifungal activity results from binding to ergosterol, a component of the cell membrane of sensitive fungi.49 This binding forms channels in the cell membrane, altering its permeability and causing leakage of Na+, K+, and H+ ions. Polyenes also bind to a lesser extent to cholesterol of mammalian plasma membrane, which accounts for most of the toxicity associated with the systemic use of amphotericin B. In addition, amphotericin B may stimulate the function of host macrophages, and this immunomodulation is mediated by the oxidized form of amphotericin B.15 Finally, amphotericin B increases the ability of C. albicans to induce the synthesis of tumor necrosis factor-α.88 Resistance to polyenes is associated with a replacement of ergosterol with other sterols in the fungal plasma membrane. A parallel decline in virulence generally occurs, however, and resistance has not been a problem clinically except for rare instances involving Candida species other than C. albicans.
Amphotericin B
Amphotericin B is an antifungal agent obtained from Streptomyces nodosus, an actinomyces found in the soil. It is a member of the polyene family of antibiotics, so called because their structure contains a large lactone (macrolide) ring with numerous conjugated double bonds (Figure 40-1). The polar hydroxylated portion and the nonpolar hydrocarbon sequence lend an amphophilic character to the molecule. Polyenes are unstable in solution because of the unsaturated chromophore region, which is easily photo-oxidized. Amphotericin B exerts either fungistatic or fungicidal activity depending on the concentration of the drug, the pH, and the fungus involved. Peak activity occurs at a pH between 6.0 and 7.5. Amphotericin B has a broad spectrum of antifungal activity and is effective against Candida species, Histoplasma capsulatum, Cryptococcus neoformans, and Coccidioides immitis.
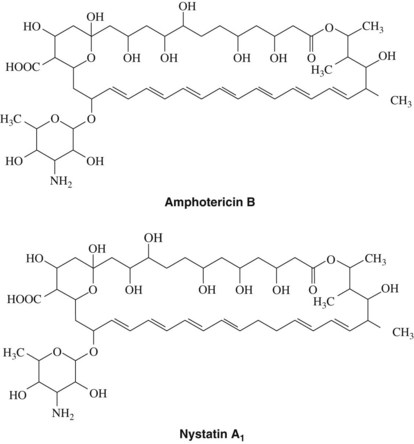
Amphotericin B is not absorbed from the skin or mucous membranes and is poorly and inconsistently absorbed from the gastrointestinal tract. Because of its insolubility in an aqueous medium, the drug is reconstituted in a solution of the bile salt deoxycholate immediately before use. For systemic infections, amphotericin B is administered by slow intravenous infusion (over 2 to 6 hours each day). The drug is bound in plasma to various lipoproteins and in tissues to cholesterol-containing membranes. More recent studies showed that amphotericin B lipid complex or liposomal amphotericin B preparations could be used for systemic infections, particularly in premature infants and other immunocompromised patients.50 Amphotericin B can also be prepared in colloidal dispersion with sodium cholesteryl sulfate in a 1 : 1 discoidal complex. Colloidal amphotericin B showed reduced peak plasma levels, prolonged residence time, and reduced renal toxicity and hepatotoxicity compared with conventional amphotericin B preparations.41
Nystatin
Nystatin is a polyene antibiotic obtained from Streptomyces noursei. Its structure is similar to the structure of amphotericin B (see Figure 40-1). Nystatin is relatively insoluble in water and unstable except as a dry powder.
Nystatin is not appreciably absorbed from the skin, mucous membranes, or gastrointestinal tract. After oral administration, the bulk of the administered dose appears unchanged in the feces. Because of unacceptable systemic toxicity, nystatin is never given parenterally. A newer form of nystatin encapsulated in liposomes showed reduced systemic cytotoxicity, however, making it an active systemic antifungal agent.38 Also, liposomal nystatin has been suggested to target Candida species that are resistant to amphotericin B.10
Nystatin is used primarily to treat candidal infections of the mucosa, skin, intestinal tract, and vagina. Although the efficacy of oral nystatin for enteric candidiasis has been questioned, topical nystatin remains a drug of choice for the treatment of candidal infections of the oral cavity (oral moniliasis, thrush, denture stomatitis). It has also been used prophylactically in immunocompromised patients.64 To treat oral candidiasis, 2 to 3 mL of a suspension containing 100,000 U/mL of nystatin is placed in the mouth, swished, and held for at least 5 minutes before swallowing. This regimen is repeated every 6 hours for at least 10 days or for 48 hours after remission of symptoms. Alternatively, 1 to 2 lozenges (200,000 U per each) may be used four to five times per day. For denture stomatitis, nystatin ointment (100,000 U/g) can be applied topically every 6 hours to the tissue surface of the denture.
Imidazole and Triazole Antifungal Drugs
Imidazoles and triazoles (together called azoles) are synthetic compounds that belong to the azole class of antifungal drugs. The antifungal spectrum of azole antifungal drugs is broad, including yeasts, dermatophytes, and various species of Histoplasma, Coccidioides, Paracoccidioides, Cladosporium, Phialophora, Blastomyces, and Aspergillus. Although the mode of action is not fully established, it is known that azoles inhibit an enzyme involved in the synthesis of fungal ergosterol. More specifically, one of the nitrogen atoms of the azole ring binds to the heme moiety of the fungal cytochrome P450 enzyme lanosterol 14-α-demethylase, inhibiting the conversion of lanosterol to ergosterol.25 The addition of ergosterol fails to reverse the antifungal effect in vitro, however, and other mechanisms must be invoked to explain the activity of these compounds against several protozoa and bacteria in which ergosterol is not an important membrane constituent. The addition of 14-α-methyl sterols such as lanosterol, whose concentrations increase as a result of azole therapy, may disrupt cell membranes even in the presence of ergosterol.
Other antifungal actions ascribed to ketoconazole and similar drugs, perhaps related to the changes caused by lanosterol, include inhibition of purine transport, interference with mitochondrial respiration, and alteration of the composition of nonsterol membrane lipids. Acquired resistance to imidazoles has not been a significant problem clinically; however, it can develop in C. albicans.82 Refractory mucosal candidiasis in immunocompromised patients has been ascribed to the Candida species with cross-resistance to clotrimazole and other azole compounds.65
Ketoconazole
Ketoconazole (Figure 40-2) is rarely used because of its toxicity and the availability of other azoles. Ketoconazole was the first oral antifungal agent to be approved for the treatment of deep systemic mycoses. It is well absorbed from the gastrointestinal tract, provided that the stomach content is acidic. Drugs that increase gastric pH, such as antacids and H2 antihistamines, markedly reduce its absorption.54 It should be reserved for cases refractory to other therapy.11,66,80
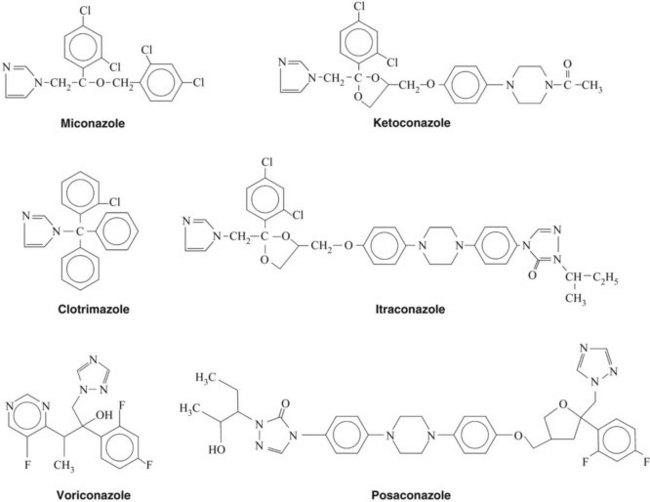
Miconazole
Miconazole (see Figure 40-2) was the first imidazole antifungal drug to be approved for topical and parenteral use. It is no longer used systemically. Cutaneous candidiasis and vulvovaginitis caused by C. albicans respond rapidly and reliably to 2% miconazole nitrate cream. Oral candidiasis is also effectively treated; however, a specific formulation for intraoral use is unavailable. Other topical uses of miconazole are treatment of cutaneous dermatophyte infections caused by Epidermophyton, Microsporum, and Trichophyton. Untoward effects after topical administration of miconazole are rare, but burning, skin maceration, itching, and redness can develop.
Clotrimazole
One troche dissolved in the mouth five times a day for 2 weeks is the standard regimen for oropharyngeal candidiasis. Patient compliance is believed to be enhanced by the more pleasant taste of clotrimazole compared with nystatin. Clotrimazole also seems to be useful for the topical treatment of oral candidiasis in patients with AIDS.80,88,89 For cutaneous candidiasis and dermatophytoses, a 1% cream or lotion is equivalent to topical miconazole.
Itraconazole
When given in therapeutic doses, itraconazole exerts effective antifungal activity against paracoccidioidomycosis, blastomycosis, aspergillosis, histoplasmosis, sporotrichosis, candidiasis, and various dermatophytoses. Previous studies show that itraconazole is effective for suppressive therapy and primary treatment of histoplasmosis in patients seropositive for human immunodeficiency virus (HIV).55,83 Drug interactions are qualitatively similar to those noted for ketoconazole, but occur less frequently. Itraconazole and related triazoles are more specific for fungal 14-α-demethylase, however, and do not affect mammalian steroid metabolism as greatly.23 Adverse effects include rashes, hepatotoxicity, hypokalemia, hypertension, and heart failure in susceptible patients.
Fluconazole
Fluconazole is active in suppressive therapy and primary treatment of cryptococcal meningitis, which may occur in patients with AIDS.46 It is effective in the treatment of mucosal candidiasis, including oropharyngeal and esophageal candidiasis.11 Weekly use of fluconazole was suggested to have prophylactic value against mucosal candidiasis in HIV-seropositive patients.71 It is also used in the primary treatment of coccidioidal meningitis and treatment of blastomycosis and histoplasmosis. In one study, fluconazole was found to be more effective against oral candidiasis than nystatin in immunocompromised children.37 It may also be effective in candidiasis resistant to polyenes and imidazoles.57
Other imidazoles and triazoles
Voriconazole and posaconazole have been developed more recently and represent a new generation of triazole antifungals with enhanced pharmacologic properties. These drugs show broad-spectrum fungicidal activity against molds and fungistatic activity against Candida and other yeasts.48 Voriconazole is a derivative of fluconazole exhibiting increased antifungal activity and specificity. It is the drug of choice for the treatment of invasive aspergillosis caused by Aspergillus terreus, which is increasingly observed as a pathogen in immunocompromised patients.74 Voriconazole is also effective against dimorphic fungi (Histoplasma, Coccidioides, and Blastomyces species), yeasts (Candida krusei, Candida glabrata, C. neoformans, and Tricosporon asahii), and pathogenic molds (Fusarium and Scedosporium species).40 Adverse side effects of voriconazole include erythematous rash, visual disturbances, hepatotoxicity, and headache.47 Voriconazole is considered a safer alternative to other antifungals such as amphotericin B for patients at risk of renal dysfunction or receiving concurrent administration of nephrotoxic drugs.81
Posaconazole is a newer addition to the antifungal triazoles that structurally resembles itraconazole (see Figure 40-2). The antifungal spectrum of posaconazole is similar to that of voriconazole except that its potential therapeutic efficacy has also been shown against Zygomycetes (e.g., Rhizopus, Absidia, and Mucor species).27 Voriconazole and posaconazole are effective against a wide variety of Candida species. In particular, voriconazole administration led to a higher success rate against C. tropicalis compared with amphotericin B and fluconazole.52 Posaconazole was found to be as effective as fluconazole for the treatment of oropharyngeal candidiasis in patients infected with HIV and led to fewer incidences of clinical relapse.79 Posaconazole may have extensive uses in dentistry in the future.
Echinocandin Antifungal Drugs
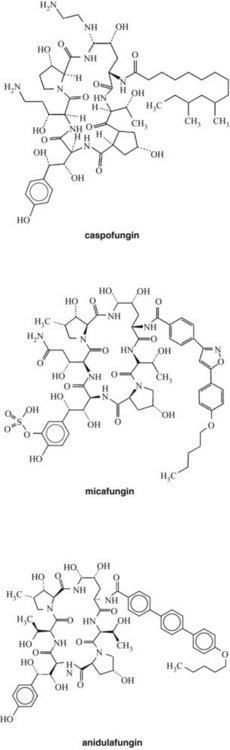
Echinocandins are a new class of antifungal drugs approved recently by the U.S. Food and Drug Administration (FDA) (Figure 40-3). Their unique mechanism of drug action involves noncompetitive inhibition of synthesis of 1,3-β-d-glucan linkages in fungal cell walls.14 The 1,3-β-d-glucan linkages are crucial for fungal cell wall synthesis and maintaining the osmotic balance. Echinocandins currently available for clinical uses include caspofungin, micafungin, and anidulafungin. Echinocandins are especially useful for treating candidal esophagitis and candidemia and Aspergillus infections, for empiric treatments of febrile neutropenia, and for antifungal prophylaxis in hematopoietic stem cell transplant (HSCT) recipients.60 Caspofungin is also approved for treatment of invasive aspergillosis in patients who are refractory to other antifungal drugs. No echinocandins are approved for pediatric patients.
Caspofungin
Caspofungin (see Figure 40-3) is derived from the fermented by-product of Glarea lozoyenisi. It is the first echinocandin approved by the FDA for clinical use. It is an echinocandin with antifungal activity against a wide variety of fungal pathogens, including Candida,8 Pneumocystis, Aspergillus, and Histoplasma species. Caspofungin disrupts the formation of the fungal cell wall by inhibiting the enzyme 1,3-β-d-glucan synthase, which is necessary for β(1,3)-d-glucan polymerization in filamentous fungi. Because this mechanism of action differs from those of amphotericin B and the azole compounds, combination therapy using caspofungin with other antifungal agents has been suggested and has yielded synergistic effects against cryptococcal species.30 Caspofungin showed higher therapeutic efficacy against candidal infections compared with amphotericin B in immunocompromised patients.15,44
Micafungin
Micafungin is a synthetic derivative of lipopeptides isolated from Coleophoma empetri. It is approved for therapeutic use against esophageal candidiasis and for chemoprophylaxis against candidiasis in neutropenic patients undergoing HSCT.60 Fluconazole has been the primary drug of choice for chemoprophylaxis against candidiasis and aspergillosis in HSCT patients. A comparative phase III clinical trial of antifungal prophylactic efficacy showed superior results, however, with micafungin compared with fluconazole.77 Among 889 adult and pediatric patients enrolled for HSCT, 50 mg of micafungin administered daily to patients yielded an 80% success rate versus 73.5% in patients who received 400 mg daily dose of fluconazole. Also, the patients receiving micafungin prophylaxis reported fewer side effects and fewer incidences of discontinued therapy compared with patients receiving fluconazole prophylaxis.
Micafungin was found to be as effective as fluconazole against esophageal candidiasis with a similar spectrum of adverse effects in HIV patients.29 A more recent study also showed a comparable level of therapeutic efficacy of micafungin against candidemia and invasive candidiasis as liposomal amphotericin B when both drugs were delivered as an intravenous infusion.53 Compared with amphotericin B, micafungin treatment led to significantly fewer adverse effects, such as hypokalemia, rigors, back pain, infusion-related reactions, and nephrotoxicity. Micafungin is considered a well-tolerated addition to the antifungal armamentarium. Micafungin is given at a daily infusion dose of 150 mg for esophageal candidiasis and 50 mg for antifungal prophylaxis.
Anidulafungin
Anidulafungin is derived from Aspergillus nidulans. It is the newest addition to echinocandin antifungals approved for esophageal candidiasis, candidemia, and invasive candidiasis. Anidulafungin showed potent and broad antifungal activity against Candida and Aspergillus species, including species resistant to fluconazole.67 Compared with azole antifungals, anidulafungin was more effective in vitro against C. albicans, C. tropicalis, C. glabrata, and C. krusei, but not Candida famata and Candida parapsilosis.9 Anidulafungin was also more effective than caspofungin against Aspergillus.68 Large-scale clinical trials confirmed the therapeutic efficacy of anidulafungin against invasive candidiasis compared with fluconazole.69 Anidulafungin is given by intravenous infusion as a 100-mg daily maintenance dose for invasive candidiasis and 50-mg daily dose for esophageal candidiasis. A loading dose is also recommended for the first day of treatment.
Other Antifungal Drugs
Flucytosine
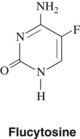
Flucytosine, a fluorinated analogue of cytosine (5-fluorocytosine) (Figure 40-4), is a synthetic antimycotic agent orally effective in the treatment of systemic fungal infections, in particular infections caused by yeasts. Flucytosine has a limited antifungal spectrum compared with amphotericin B and is mainly effective against Candida and Cryptococcus. It is also active against some species of Cladosporium and Phialophora, the latter being etiologic agents for chromoblastomycosis.
Tolnaftate and allylamine antifungal drugs
Tolnaftate is a thiocarbamate that is commonly used as a topical antifungal agent against mild-moderate superficial fungal infection in skin and toenails, such as tinea pedis, tinea cruris, tinea corporis, tinea manuum, and tinea versicolor. Susceptible dermatophytes include Malassezia furfur, Epidermophyton floccosum, Trichophyton mentagrophytes, Trichophyton tonsurans, and Microsporum canis. However, tolnaftate is generally ineffective against yeasts, however, such as C. albicans.12 Adverse effects associated with the topical use of tolnaftate are generally mild and could involve allergic contact dermatitis. Possible teratogenic effects of tolnaftate spray use during pregnancy have been suggested26 and need further investigation for conclusive results.
The mechanism of action of tolnaftate involves noncompetitive inhibition of fungal squalene epoxidase, which is a membrane-bound enzyme necessary for conversion of acetate to sterols and biosynthesis of ergosterol. This mechanism of drug action is shared by another class of antifungals known as allylamines, which include naftifine and terbinafine. Terbinafine is effective against dermatophytes (Microsporum, Trichophyton, and Epidermophyton species) and molds (Aspergillus and Scopulariopsis species).39 Terbinafine is highly lipophilic and keratophilic and accumulates in the stratum corneum of skin and nails. Similar to thiocarbamate antifungals, allylamine agents are used effectively for dermatophytosis of skin and nails. The adverse effects of terbinafine include mild and transient forms of gastrointestinal symptoms, rash, urticaria, pruritus, and neutropenia, but the drug is generally well tolerated. Terbinafine is used orally and topically, whereas naftifine is used only topically.
Griseofulvin
Griseofulvin is variably absorbed from the gastrointestinal tract; micronization of the primary drug particles (see Chapter 2) and ingestion with a fatty meal improve bioavailability. Although most of the absorbed drug is inactivated in the liver by dealkylation, the plasma half-life is fairly long (approximately 20 hours), and griseofulvin readily reaches the skin, hair, and nails, where it binds avidly to newly synthesized keratin and inhibits fungal invasion through surface keratin. Serious side effects are uncommon, but griseofulvin may induce nausea, vomiting, diarrhea, fatigue, headache, and mental confusion. The drug may also cause hematologic and dermatologic reactions. As an inducer of cytochrome P450 enzymes, griseofulvin is contraindicated in patients with acute intermittent porphyria and may participate many drug interactions, potentially decreasing the effectiveness of drugs such as warfarin and oral contraceptives. Its use has waned as a result of marketing of newer drugs for treating superficial fungal infections.
Treatment of Oral Candidiasis
Candidiasis is the most common type of oral fungal infection. Regardless of which drug is used, therapy for 2 weeks is required, and more extended treatment may be necessary. Clotrimazole, in the form of oral troches, is highly effective in most cases. On swallowing, clotrimazole can cause an increase in plasma concentrations of hepatic enzymes, which and may rarely lead to hepatitis. If patients have liver disease or are at greater risk of liver toxicity (e.g., alcoholics), nystatin oral pastilles or rinses are preferred. For more extensive disease or difficult cases, such as patients with AIDS, systemic antifungal therapy may be indicated.64,89
Oral fluconazole (100 to 200 mg/day) is a major systemic drug useful for oral candidiasis. The risk of causing liver abnormalities is less with fluconazole than the outmoded ketoconazole.23 If the infection is resistant to fluconazole, oral itraconazole (200 mg/day) is another alternative.31 Posaconazole can be administered orally at 400 mg twice daily for oral candidiasis resistant to itraconazole or fluconazole. The use of caspofungin, 50 mg intravenously, is an option in more advanced cases, as are micafungin and anidulafungin. In extreme cases, intravenous amphotericin B may be considered.31 The toxicity of this drug must be carefully weighed, and consultation with a specialist in infectious disease is essential. Surgery may be helpful to remove a condensed lesion after medical therapy. The occurrence of oral candidiasis with lichen planus is common. In these cases, a topical antifungal drug may be applied with a topical corticosteroid. It has been suggested that clotrimazole be given with a topical steroid in patients with oral lichen planus for prophylaxis against candidiasis.89 Chlorhexidine oral rinses may also be useful in treating oral candidiasis.
ANTIVIRAL AGENTS
Advances in the pharmacologic control of viral infections have lagged behind achievements in the chemotherapy of other microbial diseases. The reason for this delay, which also applies to the therapeutic management of neoplastic disorders (see Chapter 42), has been the difficulty in attaining an antiviral agent with an adequat/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


