CHAPTER 4 Pharmacogenetics and Pharmacogenomics
FDA labeling regulations now stipulate “if evidence is available to support the safety and effectiveness of the drug only in selected subgroups of the larger population with a disease, the labeling shall describe the evidence and identify specific tests needed for selection and monitoring of patients who need the drug.” So far, the FDA has recommended changes in the labeling for 6-mercaptopurine, irinotecan, and tamoxifen to include pharmacogenetic information on treatment outcome. It has also been estimated more recently that 25% of all outpatients receive at least one prescribed medication that contains pharmacogenetic information in the drug label.18 This information is not generally helpful at present because testing for most pharmacogenetic variants is not yet routine in clinical practice and most general practitioners would be incapable of using such information even if it were currently freely available. The broader field of “personalized medicine” also includes more rational targeting of drugs, such as restricting the use of trastuzumab to treat tumors based on their tumor phenotype (i.e., only those tumors that overexpress human epidermal growth factor receptor 2, the ligand for trastuzumab). This approach to therapy is sometimes included under the umbrella of pharmacogenetic testing.
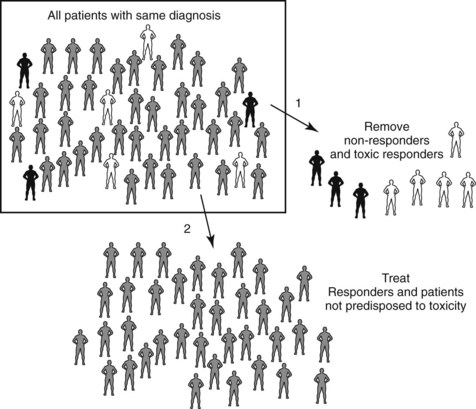
Pharmacogenetics and pharmacogenomics are areas of intense interest and development within the biotechnology and pharmaceutical industries.49 Many pharmaceutical companies are beginning to genotype patients in premarket clinical trials to exclude individuals who are predicted to experience adverse effects or therapeutic failure. This concept is illustrated in Figure 4-1. A consortium that includes major pharmaceutical companies is assembling a high-density map of the single nucleotide polymorphisms (SNPs) that exist throughout the human genome to facilitate the construction of pharmacogenetic profiles that predict drug responsiveness. SNPs occur, on average, about once every 1000 bases in the 3 billion–base human genome. A website (http://www.ncbi.nlm.nih.gov/SNP) sponsored by the National Center for Biotechnology Information maintains an updated listing of these SNPs.
This chapter presents basic principles and mechanisms that account for pharmacogenetic differences in drug therapy and toxicity. The pharmacogenetic examples included are of historical or clinical interest (or both); they are not intended to be exhaustive because, with the sequencing of the human genome, tabular listings of pharmacogenetic traits of clinical interest fall rapidly out of date. For more comprehensive and contemporary information, readers are directed to monographs,30,59 chapters,22,23 and general reviews11,13,15,25,45,61 on pharmacogenetics and pharmacogenomics, and to the regularly updated Pharmacogenomics Knowledge Base (http://www.pharmgkb.org), which forms part of the National Institutes of Health–funded Pharmacogenetics Research Network.21
Every year, more than 100,000 people die in the U.S. because they carry “misspelled” genes that make medications either ineffective or deadly. Now doctors can test for the genes before prescribing … Imagine, a lawyer asking: “Doctor, did you know this drug would kill your patient? Did you know there is a test that would have predicted that? And why did you not give your patient the test?”4
Such statements widely read by the lay public (and their lawyers) underscore the need for dental professionals to understand the role of pharmacogenetic factors in drug responsiveness. Patient malpractice claims have already alleged negligence in the use of pharmacogenetic information.50
PHARMACOKINETICS AND PHARMACODYNAMICS
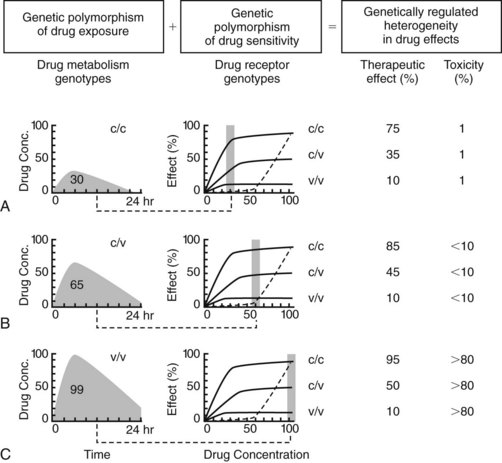
Proteins affect drug concentration (pharmacokinetics) and response (pharmacodynamics). Historically, genetic variation most often has been identified in pharmacokinetics, particularly in drug metabolizing enzymes.6 Genetic variation in the pharmacokinetic profile often necessitates a change in the dosage regimen of a drug, but not in its selection. Pharmacogenetic differences in drug target responsiveness28 are less well understood, but potentially will also have a significant impact on patient outcomes in the future. In these instances, certain drugs would be contraindicated for patients with particular genotypes. Just as drug prescribers are currently responsible for avoiding adverse “drug-drug” interactions, as described in Appendix 1, they increasingly will be held accountable for avoiding untoward “gene-drug” interactions in clinical practice. Genetic differences in pharmacokinetics and pharmacodynamics are anticipated for many, if not most, drugs, yielding important consequences for drug responsiveness, especially for agents with a narrow therapeutic index. Figure 4-2 illustrates the separate and combined influences of genetic polymorphisms in pharmacokinetics and pharmacodynamics.14
There are many gene-drug interactions with importance to dentistry. Codeine, one of the mostly commonly prescribed opioid analgesics for pain relief, is a prodrug and depends on its activation to morphine by CYP2D6, a drug-metabolizing enzyme that is known to exhibit a common genetic polymorphism in humans.48 As a result, codeine is an ineffective analgesic in a significant genetic subset (10%, depending on ethnic group) of the population. Genetic polymorphisms in opioid receptors or in second messenger systems mediating opioid receptor actions have also been observed. If a patient inherits a deficiency in CYP2D6 or the μ-opioid response system, it is unlikely that standard doses of codeine will be of therapeutic benefit. Increasing the dose of codeine to compensate for the genetic deficiency will most likely result not in analgesia but rather in an adverse reaction mediated by overstimulation of an alternative receptor that is responsive to codeine.
PHENOTYPE AND GENOTYPE
Many methods to determine the genotype have been developed in the past two decades, including restriction fragment length polymorphism analysis, allele-specific amplification, and DNA sequencing. Most methods rely on DNA amplification techniques based on the polymerase chain reaction that yield millions of copies of the specific target gene. New high-throughput methods promise to make the simultaneous determination of multiple genotypes readily available to health care professionals.52 Understanding the important and complex relationships between genotypes and phenotypes has fostered much research in functional genomics and proteomics.
ETHNIC DIFFERENCES IN PHARMACOGENETICS
The frequency of specific alleles, genotypes, and phenotypes for drug metabolizing enzymes varies widely with ethnic origin.29 A similar variance is expected for drug receptors. Clinical trials are best conducted either with ethnically diverse study populations to capture differences among ethnic groups with ethnically defined subgroups to define effects in these groups precisely. Some genotyping methods were designed originally to identify only alleles prevalent in whites. With the documented ethnic heterogeneity within the human population, however, genotyping tests need to identify all relevant alleles of a particular gene regardless of ethnic frequency.
PHARMACOGENETICS OF DRUG METABOLISM
As indicated earlier, most of the pharmacogenetic traits identified to date occur in genes encoding drug metabolizing enzymes. It is anticipated that genetic polymorphisms may be identified in all drug metabolizing enzymes. Many of these genetic polymorphisms are already known to be important in therapeutics,63 and examples of historical and clinical interest are highlighted next.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


