CHAPTER 39 Antibacterial and Antibiotic Drugs*
OROFACIAL INFECTIONS
Oral Microbial Pathogens and Associated Oral Infections
Acute orofacial infections
Table 39-1 presents quantifiable data from 12 clinical studies from 1976-1996 on the microbiology of acute orofacial infections. The average number of isolates per case was 3.6, with a maximum of 12. The data in Table 39-1 indicate that acute orofacial infections are polymicrobial, dominated by anaerobes, and often contaminated by various microorganisms, particularly from the pharynx, sinuses, and gastrointestinal tract.
TABLE 39-1 Microorganisms Associated with Acute Orofacial Abscesses Based on 2339 Isolates in 12 Studies from 1976-1996
| MICROORGANISM | NO. OF ISOLATES | PERCENT OF TOTAL |
|---|---|---|
| Aerobes/Facultative | ||
| VGS | 470 | 20.1 |
| Staphylococcus aureus/Staphylococcus epidermidis | 136 | 5.8 |
| β-Hemolytic streptococci | 68 | 2.9 |
| Total | 674 | 28.8 |
| Anaerobes | ||
| Prevotella/Porphyromonas* | 641 | 27.4 |
| Peptostreptococcus | 388 | 16.6 |
| Fusobacterium | 181 | 7.7 |
| Eubacterium | 87 | 3.7 |
| Veillonella | 58 | 2.5 |
| Actinomyces | 47 | 2 |
| Total | 1402 | 9.8 |
Each of the following species is less than 1% of total but together constitute 11.3% of all isolates: Acinetobacter; Aggregatibacter actinomycetemcomitans; Arachnia; Citrobacter; Corynebacterium; Eikenella corrodens; Clostridium; Enterobacter; Escherichia coli; group A, B, C, D, and G streptococci; Haemophilus influenzae; Klebsiella pneumoniae; Lactobacillus; Neisseria; Propionibacterium acnes; Serratia; and spirochetes (most likely contaminants).
VGS, Viridans group streptococci.
* Most studies list as Bacteroides.
Metastatic infections from Porphyromonas gingivalis, Prevotella intermedia, and Prevotella nigrescens and other anaerobic periodontal pathogens apparently are very rare,68,83 whereas respiratory tract infections may commonly precede pericoronitis. Staphylococci isolated from facial cellulitis are most likely contaminants because these organisms are not a normal component of the subgingival flora residing primarily on oral mucosal surfaces. They can be a major factor in the cause of oral mucositis. Anterior nares carriage of Staphylococcus aureus occurs permanently in 20% and intermittently in 60% of the population.53 Subgingival staphylococci may appear because of selection by local or systemic antibiotic therapy.23 Retropharyngeal abscesses seem to have the same microbial cause as facial cellulitis.
Pulpal and periapical pathogens
Commonly isolated microorganisms associated with pulpal/periapical lesions seem to be VGS, other streptococci (β-hemolytic, β-hemolytic, group D), Fusobacterium, Peptostreptococcus micros, Lactobacilli, Actinomyces, Porphyromonas, Prevotella, Veillonella, Eubacterium, and Bacteroides forsythus. Other microorganisms found less commonly or rarely include Propionibacterium acnes, Candida albicans, Enterococcus, staphylococci, Pseudomonas aeruginosa, Enterobacter aerogenes, Serratia marcescens, Eikenella corrodens, S. pneumoniae, Corynebacterium, Capnocytophaga, Selenomonas, and Wolinella (Box 39-1). A significant portion of these may be contaminants.
Periodontal abscesses
An acute periodontal abscess is characterized as a lesion of periodontal breakdown located within the gingival wall of the periodontal pocket and manifested as a localized accumulation of purulence.40 An acute periodontal abscess may result from an exacerbation of local periodontitis pathology, after periodontal debridement procedures, or from the lodgment of a foreign object in the periodontal pocket (e.g., popcorn husks, dental floss, calculus).40 The microbial cause of a periodontal abscess is similar to that of adult periodontitis, and the flora is commonly indistinguishable from the microflora of the subgingival plaque in adult periodontitis.40 The predominant microflora are P. gingivalis (55% to 100% of isolates), P. intermedia (25% to 100% of isolates), Fusobacterium nucleatum (44% to 65% of isolates), Aggregatibacter actinomycetemcomitans (25% of isolates), Campylobacter rectum (80% of isolates), Prevotella melaninogenicus (22% of isolates), and Treponema denticola (71% of isolates)40; other organisms include P. micros and B. forsythus.40 It has been estimated that 74% may be anaerobes and 67% may be gram-negative rods, with streptococci significant only at the base of the abscess.73
The principal and sometimes only therapy of periodontal abscess is incision and drainage through the external tissue and compression of the soft tissue wall.40 Curettage or root planing is not usually required unless a reasonable chance exists to eliminate the periodontal pocket.40 The abscess tends to become fistulous readily and rarely results in metastasis or acute orofacial cellulitis, possibly because VGS have been replaced by periodontal pathogens that do not spread by fascial planes as do streptococci. On fistulization, the lesion is self-limiting, as opposed to dentoalveolar abscesses of pulpal origin, which may readily end in cellulitis.
Periodontal abscess often can be treated simply with incision and drainage without antibiotics because it is rarely associated with fever, malaise, lymphadenopathy, and other signs of systemic involvement; periodontal abscess may necessitate antibiotic therapy only if the signs and symptoms of systemic involvement or cellulitis are present, or incision and drainage cannot be performed.40,60,80 This is in contrast to antibiotic therapy of pulpal/periapical infection, which should be more aggressive because there is a much greater tendency to spread into the fascial planes. If antibiotic therapy of a periodontal abscess is indicated, the situation is classic for short-term, high-dose therapy, as opposed to commonly longer therapy for dentoalveolar abscess.60,81 Periodontal pathogens rarely, if ever, metastasize to the heart or other organs and tissues.82,83
Acute necrotizing ulcerative gingivitis
The microbiology of acute necrotizing ulcerative gingivitis (trench mouth, Vincent’s infection) is characterized primarily by Treponema, Fusobacterium, Selenomonas, and P. intermedia and secondarily by Veillonella, Neisseria, Capnocytophaga, E. corrodens, Bacteroides, Actinomyces, and gram-positive cocci.65
Pericoronitis
The microbial flora of pericoronitis are a complex mixture of organisms resembling that of periodontitis and gingivitis,58 often with a high concentration of VGS. Common microorganisms found in 40% or more of samples include Stomatococcus, Rothia dentocariosa, Actinomyces naeslundii, Actinomyces israelii, Prevotella, Neisseria, Haemophilus, P. micros, Capnocytophaga, Corynebacterium, Bifidobacteria, and treponemes.58 Other, less common isolates include coagulase-negative staphylococci (CoNS), lactobacilli, Veillonella, Fusobacterium, and Porphyromonas.58
Periimplantitis
The microbial causes of chronic periodontitis, refractory periodontitis, and periimplantitis (an inflammatory process of the tissues surrounding an osseointegrated implant resulting in loss of supporting bone) are remarkably similar, differing primarily in the quantitative and not qualitative isolation of the predominant species: B. forsythus, F. nucleatum, P. gingivalis, P. intermedia, P. nigrescens, C. rectum, and treponemes (spirochetes).62 Healthy dentulous or implant periodontium usually exhibits fewer of the above-mentioned organisms and is dominated by VGS, Actinomyces, Veillonella, E. corrodens, and Capnocytophaga.57
Periimplantitis results from a shift in periodontal flora with facultative anaerobic streptococci (VGS) and nonmotile rods replaced by gram-negative anaerobic bacilli and spirochetes, similar to what occurs in periodontitis.57,62 The issue of whether implant success is compromised in patients with periodontitis (treated or untreated) is controversial, but it seems reasonable to postulate that the gingival sulcus in patients with periodontitis is a reservoir for periodontal microbial pathogens.84 Host resistance and factors that reduce immunity (stress) are as likely a complicating factor in periimplantitis as they are in aggressive periodontitis.
Osteomyelitis
Osteomyelitis is an infectious inflammatory process resulting in bone destruction. There are several clinical descriptions of osteomyelitis as follows: (1) secondary to a contiguous infection, (2) secondary to vascular deficiency and diabetic foot infection, (3) in association with an infected prosthesis (e.g., dental implant, prosthetic joint), (4) hematogenous, (5) chronic, and (6) acute.21,59 Osteomyelitis, resulting from local spread from a contiguous contaminated or infectious source, commonly follows trauma, bone surgery, or joint replacement. Approximately 15% of diabetics develop foot pathology requiring amputation with contributing factors of bone and soft tissue ischemia and peripheral motor, sensory, and autonomic neuropathy. The amount of osteomyelitis associated with various bone implants is increasing; this includes dental implants.21,59 Spread of bacteria to bone via the hematogenous route is particularly common in prepubertal children and immunocompromised elderly patients. Chronic osteomyelitis is a long-standing infection of months to years in duration characterized by persistent microorganism, low-grade inflammation, sequestrum formation, and fistulous tracts. Acute osteomyelitis evolves over a few days or weeks.
The treatment of osteomyelitis usually involves antibiotic therapy and surgery. Because of the multiplicity of etiologic organisms, it seems imperative to get a culture and sensitivity test as soon as possible to initiate the most appropriate antibiotic therapy.45 The disease may have a polymicrobial etiology with an average of 3.9 microbes per culture. The microbial cause of oral osteomyelitis varies to some extent with the anatomic site. The most common microorganisms in orofacial osteomyelitis include streptococci, lactobacilli, Eubacterium, Klebsiella pneumoniae, S. aureus, Acinetobacter baumanii, and P. aeruginosa.21
Deep neck space infections
Deep neck space infections arise most commonly from upper airway infections (47.5% to 53.2%) and odontogenic infections (28.8% to 30.5%).10,46 The spaces involved include the sublingual, submylohyoid (submental, submandibular), lateral pharyngeal (parapharyngeal, peripharyngeal, pterygopharyngeal, retropharyngeal), and masseter spaces (masseter, pterygotemporal, parotid, peritonsillar).87 These infections include Lemierre’s syndrome (suppurative thrombophlebitis of the internal jugular vein from septic emboli) and Ludwig’s angina. Deep neck infections seem to be more common in patients with diabetes mellitus47 and patients with low socioeconomic status and with poor oral hygiene.3
The microbiology of deep neck space infections is primarily that of commensal microorganisms that for some reason travel quickly through the fascial planes, which likely have acquired virulence genes they do not normally possess. These organisms include VGS, S. aureus, Peptostreptococcus, K. pneumoniae, and oral anaerobes (Porphyromonas, Prevotella, and Fusobacterium species).9
There are no data to support pretreatment or postsurgical antibiotic prophylaxis to prevent deep neck space infections (see Chapter 49). For unknown reasons, these normally commensal and harmless bacteria become very aggressive and spread rapidly to the submandibular regions. These infections are often polymicrobial, making antibiotic treatment alone very difficult, and most must be treated by incision and drainage.
Ludwig’s angina
As first described by Ludwig in 1836, Ludwig’s angina is characterized by a massive bilateral edema of the mouth floor with pathognomonic elevation of the tongue against the palate and posterior pharyngeal wall along with glottic edema, resulting in potentially life-threatening airway obstruction—hence the vernacular terms morbus strangulatoris, angina maligne, and garotillo (“hangman’s noose”). Ludwig’s angina involves the connective tissues, fascia, and muscle and spreads by fascial planes through the submandibular, sublingual, and submental spaces and potentially on to the pharynx, retropharyngeal region, and mediastinum.77 Approximately 70% to 80% of cases are of odontogenic origin, with 99% exhibiting bilateral swelling in the neck; 95%, an elevated tongue; 89%, fever; and 51%, trismus.77 In 71 patients in whom cultures were obtained, 35% of species were VGS, 28% were “other” streptococci, 14% were staphylococci, and 27% were Porphyromonas and Prevotella and other anaerobes, with a few isolates of P. aeruginosa, K. pneumoniae, H. influenzae, S. pneumoniae, and Escherichia coli.70 The heavy preponderance of streptococci emphasizes the ability of these organisms to move through tissue rapidly.
Necrotizing fasciitis
Necrotizing fasciitis is a rare but often fatal infection involving the superficial fascial layers of the neck, extremities, abdomen, and perineum.36 It was first described by Hippocrates and has also been known as streptococcal, hospital, or galloping gangrene. Most recently, the lay press has labeled it the “flesh-eating disease.” The term necrotizing fasciitis was first employed in 1952.36
The most common cause of head and neck necrotizing fasciitis is dental infection (odontogenic origin) with 9% of all cases located in the head and neck region.38 In a review of 125 literature cases, it was found that (1) the male/female ratio was 3 : 1; (2) the origin of the infection was 66 in the mandible, 11 in the maxilla, and 48 nondelineated; (3) periapical infection was the most common cause; and (4) 70 of 125 patients had systemic complications (e.g., alcoholism, hypertension, liver cirrhosis, acquired immunodeficiency syndrome [AIDS], intravenous drug abuse, and renal insufficiency). Despite aggressive therapy, the mortality rate was 19.2%.103 In other scenarios, death may occur within hours, and the mortality rate may be 50%.
Microbial Resistance in Orofacial Pathogens
In the 1970s, viridans and anaerobic streptococci were universally sensitive to the β-lactams, with 90% to 99% also sensitive to erythromycin and clindamycin. In 1983, a high rate of penicillin resistance in VGS was detected in South Africa in the oral flora of children with a similar high penicillin resistance in pneumococci.35 Currently, β-lactamase enzymes are common in oral microorganisms, and VGS (Streptococcus milleri, Streptococcus mutans, Streptococcus salivarius, Streptococcus sanguis, Streptococcus mitis groups) with altered penicillin binding proteins (PBPs) are increasingly resistant to β-lactams and macrolides.
In children treated for otitis media and exposed to repeated antibiotics who had samples taken of their supragingival plaque, 60% of S. sanguis isolates were resistant to at least one antibiotic; 26%, to at least two antibiotics; 32%, to amoxicillin; 24%, to penicillin V; and 20%, to amoxicillin and penicillin V.31 In 139 cultures of VGS isolated from mixed orofacial infections, 23% were resistant to penicillin G; 45%, to erythromycin; 46%, to clindamycin; and 44%, to levofloxacin; 100% were sensitive to minocycline.55 Reports of 23% to 81% resistance rates of VGS to ampicillin and amoxicillin in hospitalized patients and patients in the community are common, depending on the breakpoint chosen for resistance.
In a cohort of Japanese children at high risk for bacterial endocarditis, 31.7% of VGS exhibited resistance at MICs of 4 to 16 µg/mL.74 Children treated with long-term penicillin for the prevention of rheumatic fever were found to have resistance rates of 78% to 81%. The problem is compounded further because many oral streptococci are resistant to multiple antibiotics with reduced sensitivities to cephalosporins, macrolides, and clindamycin,55 as shown by a Taiwan study reporting a 20% to 50% resistance rate to clindamycin and 30% to 70% resistance to tetracycline in penicillin-resistant Streptococcus oralis.99
In the United States, 40% to 50% of sampled VGS are resistant at MICs greater than or equal to 0.25 µg/mL,49 whereas in a study of 43 U.S. medical centers from 1993-1994, 352 VGS blood cultures exhibited a resistance rate of 13.4% at MICs greater than or equal to 4 µg/mL (high resistance) and 42.9% at MICs of 0.25 to 2 µg/mL (intermediate resistance).28 Intermediate resistance commonly escalates to high resistance over time.
Of considerable concern is the high β-lactam resistance rate in VGS in patients with neutropenia associated with hematologic malignancies and patients at risk for infective endocarditis. Approximately 18% to 21% of bacteremias experienced by immunocompromised patients may be caused by VGS, particularly S. mitis with a 3.2% to 40% resistance rate to penicillin G and cephradine,86 with some at MICs of 0.25 to 4 µg/mL for penicillin and 2 to 32 µg/mL for cephradine.
β-Lactamase production is also present in oral Veillonella, Fusobacterium, Capnocytophaga, P. aeruginosa, and B. forsythus. Lengthy or repeated antibiotic exposure increases the presence of β-lactamase in oral Prevotella, Porphyromonas, and Fusobacterium.41,76 Resistance genes may be shared between family members.54
Resistance to fluoroquinolones in VGS is increasing, and the resistance factors can be transferred between VGS, Streptococcus constellatus, and S. pneumoniae, providing for efflux mechanisms or point mutations in topoisomerase IV or DNA gyrase. Methicillin-resistant S. aureus (MRSA) may be present in the oral cavity of children for 5 or more years.97
In a study of gingival crevicular fluid microorganisms found in periodontitis patients and their sensitivities to seven antibiotics at two different time periods (1980-1985 and 1991-1995), the resistance rates increased by 172% to tetracycline, 193% to doxycycline, 133% to penicillin G, 238% to amoxicillin, 116% to erythromycin, and 108% to clindamycin.107 VGS showed variable sensitivities: 85% to 100% to penicillin G, 75% to 100% to amoxicillin, 46% to 100% to clindamycin, and 34% to 74% to tetracyclines.
ANTIBACTERIAL ANTIMICROBIAL DRUGS
Antibacterial drugs are primarily classified according to their chemical class and mechanism of action.5 They also can be distinguished based on spectrum and adverse effects. In addition to these aspects of antimicrobial drugs, the therapeutic uses, including dental applications, of each class of drugs are discussed.
β-Lactam Antibiotics
Penicillin has been said to have “brought more curative power to a barefoot, itinerant care provider in the deepest reaches of Africa than the collective powers of all the physicians in New York City.”69 Yet with the seemingly infinite ability of humans to push a system until it breaks, we now have a multiplicity of microorganisms that were initially exquisitely sensitive to the antimicrobial effects of penicillins but that are now highly resistant to their killing power. β-Lactam antibiotics remain the most widely used antibiotics in the world, however, because of their broad spectrum of activity and relative lack of toxicity despite a relatively high incidence of allergy.
Penicillins
Chemistry and classification
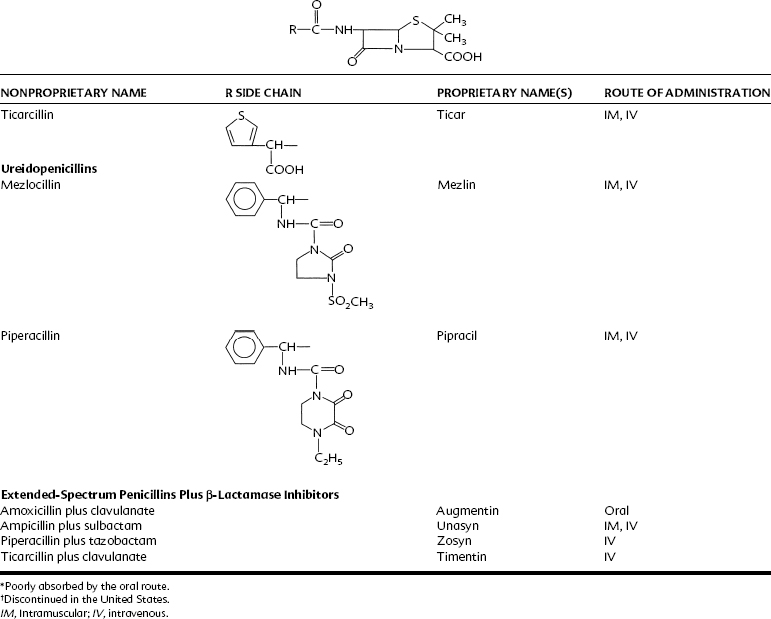
Penicillin is a cyclic dipeptide consisting of two amino acids (d-valine, l-lysine), a particular molecular configuration unknown in higher life forms (Figure 39-1). The synthesis in 1958 of the basic structure of penicillins (6-aminopenicillanic acid) allowed for its manipulation by the addition of various side chains to the β-lactam and thiazolidine rings. Different salts (Na+, K+, procaine, benzathine) were also created for pharmacokinetic purposes. On the basis of these modifications, penicillins can be divided into four groups: penicillin G and its congeners, β-lactamase–resistant (stable) penicillins, extended-spectrum penicillins, and extended-spectrum penicillins with β-lactamase inhibitors (Table 39-2).
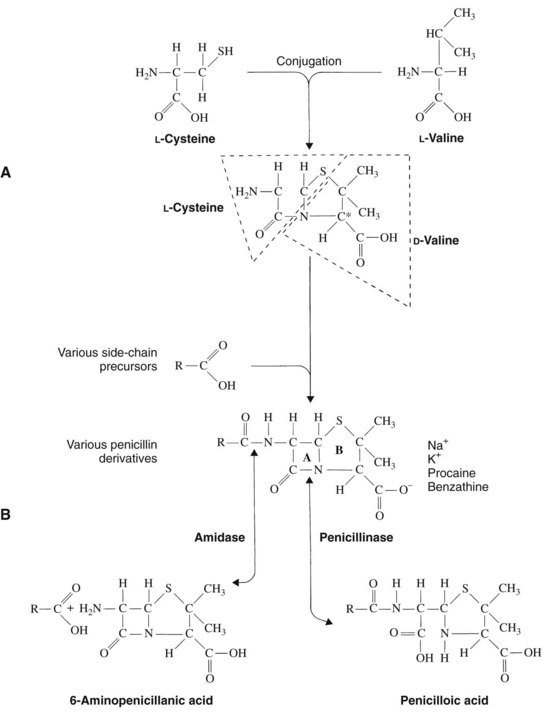
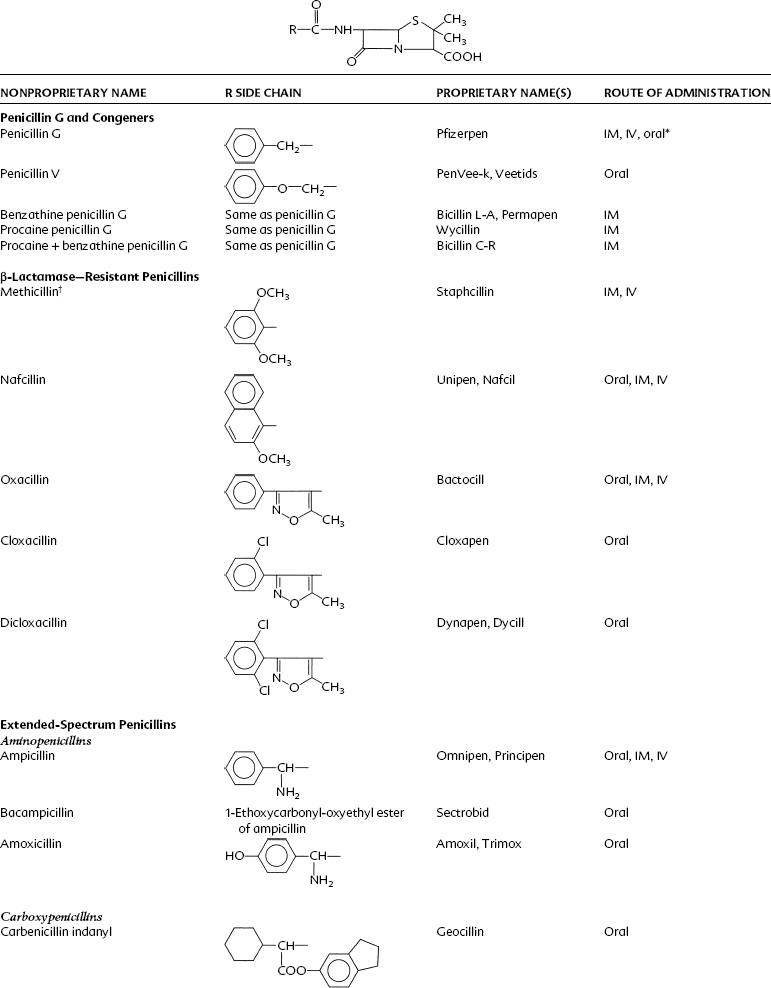
Acid-stable penicillins are resistant to breakdown in stomach acid, indicating their usefulness as oral drugs. Penicillin V, amoxicillin, and cloxacillin are examples. Other examples of orally useful drugs are listed in Table 39-2. Penicillinase-resistant penicillins are resistant to some β-lactamases. Bacteria, particularly staphylococci, develop resistance to penicillins chiefly through the elaboration of β-lactamase enzymes (penicillinases) that inactivate the penicillins by cleavage of the 6-aminopenicillanic acid nucleus to yield penicilloic acid derivatives. The production of staphylococcal penicillinase is encoded in a plasmid and may be transferred to other bacteria. Methicillin was the first semisynthetic derivative to be introduced that was stable in the presence of β-lactamase. Subsequently, nafcillin and three isoxazolyl derivatives (oxacillin, cloxacillin, and dicloxacillin) were marketed. The structural formulas for these semisynthetic derivatives are shown in Table 39-2.
Extended-spectrum penicillins are represented by two groups of penicillin derivatives. One group includes ampicillin, the first extended-spectrum penicillin to be introduced; amoxicillin, a close congener of ampicillin; and bacampicillin, a drug that is rapidly hydrolyzed in vivo to yield ampicillin (which accounts for its pharmacologic and toxicologic effects). The second group contains carbenicillin, the first penicillin to exhibit activity against Pseudomonas and indole-positive Proteus species, and ticarcillin, mezlocillin, and piperacillin, drugs with improved activity against P. aeruginosa.75 The molecular structures of these agents are depicted in Table 39-2. Carbenicillin for injection is no longer available in the United States, but carbenicillin indanyl, the oral form, is still used.
Mechanism of action and antibacterial spectrum
Penicillins as agents of choice in treating specific organisms are listed in Box 39-2 (primarily according to the Medical Letter of Drugs and Therapeutics).18,33,110 Amoxicillin and penicillin V are drugs of choice against VGS, Peptostreptococcus, E. corrodens, F. nucleatum, A. israelii, Clostridium tetani, C. perfringens, Leptotrichia buccalis, Neisseria, and non–β-lactamase–producing Prevotella and Porphyromonas.18,33,110 Amoxicillin plus clavulanate is additionally effective against K. pneumoniae, Enterobacter, M. catarrhalis, Bacteroides fragilis, non–methicillin-resistant and β-lactamase–producing staphylococci, and β-lactamase–producing Prevotella and Porphyromonas.33
Amoxicillin and penicillin V are the initial drugs of choice in orofacial infections in nonallergic patients but are ineffective against streptococci (VGS) with altered PBPs. The clinical impact of antibiotic failures against these resistant streptococci and gram-negative β-lactamase–producing oral anaerobes is likely to be significant but has yet to be determined by clinical studies. On the basis of the antimicrobial spectrum of penicillin G and V and other clinical characteristics, the drugs are useful in the treatment of numerous diseases (Box 39-3).
Absorption, fate, and excretion
Table 39-3 lists important pharmacokinetic properties of oral penicillins.13,72 Penicillin G (benzylpenicillin) is rarely used orally because of its poor gastric absorption rate. If it is prescribed orally, it should be given at doses four to five times greater than drugs used parenterally. Penicillin V and amoxicillin are well absorbed orally, with amoxicillin considerably superior in its half-life and peak serum concentrations. Better oral absorption argues for the use of amoxicillin over penicillin V, but both drugs are effective in microorganism-sensitive orofacial infections and are equally inactive against VGS with altered PBPs.
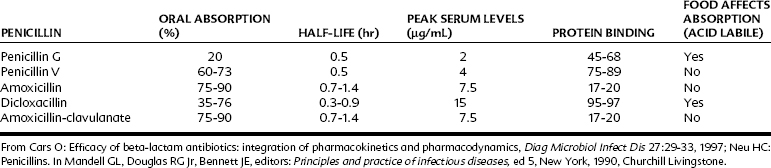
Procaine penicillin G and benzathine penicillin G are repository forms prepared for intramuscular injection with slow release from the injection site (Figure 39-2). The free non–protein-bound serum concentrations of penicillins are 0.9 µg/mL for penicillin G, 0.8 µg/mL for penicillin V, 0.45 µg/mL for dicloxacillin, and 6.2 µg/mL for amoxicillin.13 The route of excretion is primarily by the kidneys, with limited liver metabolism.13
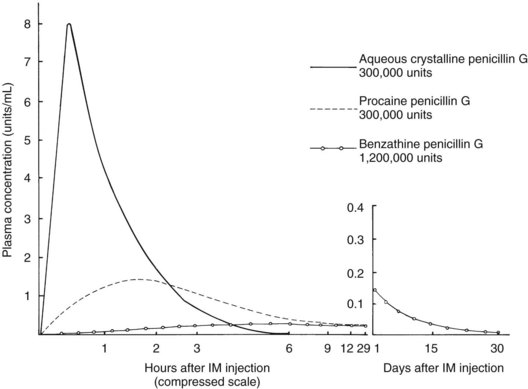
β-Lactam antibiotics produce time-dependent killing of bacteria, and frequent dosing is required to maintain relatively constant blood levels with as little fluctuation as possible.101 The killing power of β-lactams is maximum at three to four times the MIC of susceptible microorganisms.101 The prime determinant of the efficacy of β-lactams is the length of time the concentration of the drug in the infected area is greater than the MIC of the infecting organism.101
To be maximally effective, the serum and tissue concentrations of β-lactams should be greater than the MIC for 50% to 70% of the dosing interval.22 The current package insert recommends dosing intervals of 6 hours for penicillin V and first-generation oral cephalosporins. Some drugs have very short half-lives (30 to 45 minutes),33 and consequently 6-hour dosing intervals may result in very low serum levels in the last 2 or 3 hours. Continuous intravenous penicillin is receiving greater attention as a way to circumvent this problem.
β-Lactamase inhibitors
Currently, three agents are available to bind irreversibly to the catalytic site of susceptible β-lactamases to prevent hydrolysis of β-lactam antibiotics: clavulanic acid, sulbactam, and tazobactam. Clavulanic acid is derived from Streptomyces clavligerus, sulbactam is a semisynthetic penicilloic acid sulfone, and tazobactam is chemically related to sulbactam.11 All β-lactamase inhibitors have the same mechanism of action, which is to bind to the active site of β-lactamases, where they are converted to an inactive product by β-lactamase (“suicide inhibition”).11 Only clavulanic acid is orally absorbed. Clavulanic acid is combined with amoxicillin, sulbactam with ampicillin, and tazobactam with piperacillin.
The sole therapeutic use of β-lactamase inhibitors is to prevent the hydrolysis of penicillins in the management of β-lactamase–producing microorganisms responsible for otitis media and sinusitis (S. pneumoniae, H. influenzae, M. catarrhalis), nosocomial pneumonia (MSSA or K. pneumoniae), intra-abdominal abscesses from β-lactamase–producing anaerobes and other microorganisms, and some upper respiratory tract infections. β-lactam/β-lactamase inhibitor combinations offer no advantage against non–β-lactamase–producing microorganisms and are ineffective against MRSA; many CoNS and enterococci; and the inducible β-lactamases produced by P. aeruginosa, S. marcescens, Enterobacter cloacae, Citrobacter freundii, and M. morganii.11 These β-lactam/β-lactamase inhibitor combinations can often be useful as alternative antibiotics against Bacteroides, M. catarrhalis, E. coli, K. pneumoniae, indole-positive Proteus, Providencia rettgeri, Providencia stuartii, E. corrodens, Pasteurella multocida, and Pseudomonas pseudomallei (see Box 39-2).18
Therapeutic uses in dentistry
In some instances, penicillins G and V and amoxicillin are unsuitable for treating oral infections. Some dental infections are caused by β-lactamase (penicillinase)–producing organisms, and in such cases the appropriate antibiotic is a penicillinase-resistant penicillin derivative, erythromycin, or clindamycin. Patients who have been receiving extended prophylactic therapy with penicillin for the prevention of rheumatic fever generally require another antibiotic if they acquire an infection or require endocarditis prophylaxis. Certain periodontal infections are associated with gram-positive and gram-negative aerobic and anaerobic microorganisms, for which an antimicrobial agent with a more extended antibacterial spectrum, such as amoxicillin or more commonly a β-lactam/β-lactamase agent combined with metronidazole, may be the agent of choice. Table 39-4 summarizes antimicrobial therapy based on pathogens; it emphasizes the importance of penicillin V and amoxicillin.
TABLE 39-4 Drugs Used to Treat Infections Caused by Specific Microorganisms
| MICROORGANISM | DRUG OF FIRST CHOICE | ALTERNATIVE DRUGS* |
|---|---|---|
| Gram-Positive Cocci | ||
| Staphylococcus species | ||
| Methicillin-sensitive | Penicillinase-resistant penicillin (e.g., cloxacillin) | First-generation cephalosporin, vancomycin, clindamycin, imipenem, meropenem, luoroquinolone, linezolid, daptomycin |
| Methicillin-resistant | Vancomycin with or without gentamicin or rifampin | Quinupristin-dalfopristin, linezolid, fluoroquinolone, doxycycline, trimethoprim-sulfamethoxazole, tigecycline, daptomycin |
| Streptococcus pyogenes | Penicillin G or V | Cephalosporin, erythromycin, vancomycin, clindamycin, clarithromycin, azithromycin, linezolid, daptomycin |
| Streptococcus viridans group | ||
| Oral infections | Penicillin G or V | Erythromycin, clindamycin, cephalosporin |
| Bacteremia or endocarditis | Penicillin G with or without gentamicin | Ceftriaxone, vancomycin |
| Streptococcus, anaerobic (Peptostreptococcus) | Penicillin G or V | Cephalosporin, clindamycin, vancomycin |
| Streptococcus pneumoniae | Penicillin G or V, amoxicillin | Cephalosporin, trimethoprim-sulfamethoxazole, erythromycin, clindamycin, clarithromycin, azithromycin, levofloxacin, gemifloxacin, moxifloxacin, meropenem, imipenem, ertapenem |
| Enterococcus species | Ampicillin, amoxicillin, penicillin G with gentamicin | Vancomycin with gentamicin, or linezolid, quinupristin-dalfopristin |
| Gram-Negative Cocci | ||
| Neisseria gonorrhoeae | Ceftriaxone | Cefotaxime, cefixime, penicillin G |
| Neisseria meningitidis | Penicillin G | Cefotaxime, ceftizoxime, ceftriaxone, chloramphenicol, fluoroquinolone, sulfonamide |
| Moraxella (Branhamella) catarrhalis | Fluoroquinolone, cefuroxime | Trimethoprim-sulfamethoxazole, amoxicillin-clavulanate, erythromycin, clarithromycin, azithromycin, doxycycline, cefotaxime, ceftizoxime, ceftriaxone |
| Gram-Positive Bacilli | ||
| Bacillus anthracis | Ciprofloxacin, tetracycline | Penicillin G, erythromycin, amoxicillin, imipenem, clindamycin, levofloxacin |
| Clostridium difficile | Metronidazole | Vancomycin |
| Clostridium perfringens | Penicillin G, clindamycin | Imipenem, meropenem, ertapenem, metronidazole, chloramphenicol |
| Clostridium tetani | Metronidazole | Penicillin G, doxycycline |
| Corynebacterium diphtheriae | Macrolide | Penicillin G |
| Corynebacterium species (diphtheroids) | Vancomycin or penicillin G with gentamicin | Erythromycin |
| Gram-Negative Bacilli | ||
| Bacteroides, oropharyngeal strains | Penicillin G | Cefotetan, cefoxitin, clindamycin, metronidazole, ampicillin-sulbactam, amoxicillin-clavulanate |
| Capnocytophaga canimorsus | Penicillin G or V | Cefotaxime, ceftriaxone, ceftizoxime, clindamycin, ciprofloxacin, imipenem, meropenem, vancomycin, fluoroquinolone |
| Eikenella corrodens | Ampicillin, amoxicillin-clavulanate | Erythromycin, ceftriaxone, doxycycline, clarithromycin, azithromycin |
| Escherichia coli | Cefotaxime, ceftizoxime, ceftriaxone, ceftazidime, cefepime | Ciprofloxacin, ampicillin with or without gentamicin, tobramycin or amikacin, aztreonam, extended-spectrum penicillin with penicillinase inhibitor, trimethoprim-sulfamethoxazole, imipenem, meropenem, ertapenem, fluoroquinolone, tigecycline |
| Fusobacterium species | Penicillin G, penicillin V, metronidazole | Clindamycin, cefoxitin, erythromycin |
| Haemophilus influenzae | Cefotaxime, ceftriaxone, trimethoprim-sulfamethoxazole | Ampicillin or amoxicillin with or without penicillinase inhibitor, cefaclor, cefuroxime, fluoroquinolone, clarithromycin, azithromycin |
| Klebsiella pneumoniae | Cefotaxime, ceftriaxone, ceftazidime, cefepime | Aminoglycoside, aztreonam, fluoroquinolone, imipenem, meropenem, ertapenem, mezlocillin, extended-spectrum penicillin with penicillinase inhibitor, piperacillin, tigecycline, trimethoprim-sulfamethoxazole |
| Legionella pneumophila | Azithromycin or ciprofloxacin or other fluoroquinolone with or without rifampin | Trimethoprim-sulfamethoxazole, erythromycin, doxycycline, with or without rifampin |
| Leptotrichia buccalis | Penicillin G, penicillin V | Clindamycin, doxycycline, erythromycin |
| Proteus mirabilis | Ampicillin | Aminoglycoside, cephalosporin, fluoroquinolone, ticarcillin/clavulanate, piperacillin/tazobactam, aztreonam, imipenem, meropenem, ertapenem |
| Pseudomonas aeruginosa | Ticarcillin/clavulanate, piperacillin/tazobactam with or without aminoglycoside, ciprofloxacin | Aztreonam, ceftazidime, cefepime or imipenem with aminoglycoside, doripenem |
| Salmonella typhi | Ceftriaxone, ciprofloxacin, or levofloxacin | Amoxicillin, ampicillin, trimethoprim-sulfamethoxazole, chloramphenicol |
| Shigella | Fluoroquinolone | Trimethoprim-sulfamethoxazole, ampicillin, azithromycin, ceftriaxone |
| Other Microorganisms | ||
| Mycobacterium tuberculosis | Isoniazid with rifampin (or rifabutin or rifapentine) and pyrazinamide with or without ethambutol | Ethambutol, streptomycin, amikacin, ciprofloxacin, ofloxacin, capreomycin, kanamycin, ethionamide, aminosalicylic acid, cycloserine (in combinations) |
| Actinomyces israelii | Penicillin G | Doxycycline, erythromycin, clindamycin |
| Nocardia asteroides | Trimethoprim-sulfamethoxazole | Minocycline, sulfisoxazole, amikacin, imipenem, meropenem, ceftriaxone, linezolid, cycloserine |
| Treponema pallidum | Penicillin G | Ceftriaxone, doxycycline |
| Chlamydia psittaci | Doxycycline | Chloramphenicol |
| Rickettsiae | Doxycycline | Fluoroquinolone, chloramphenicol, rifampin |
| Candida albicans | ||
| Oral lesions | Nystatin, clotrimazole, fluconazole | Itraconazole |
| Systemic infections | Fluconazole, itraconazole | Amphotericin B with or without flucytosine |
| Viruses† | ||
| Herpes simplex | ||
| Orolabial | Penciclovir | |
| Keratitis | Acyclovir | Trifluridine, foscarnet |
| Genital infection | Acyclovir | Valacyclovir, famciclovir |
| Encephalitis | Acyclovir | |
| Human immunodeficiency virus | Zidovudine with another nucleoside analogue, plus a protease inhibitor† | Zidovudine with another nucleoside analogue, plus nevirapine, with or without fusion inhibitor |
| Influenza A | Rimantadine | Amantadine, zanamivir, oseltamivir |
Adverse effects
The adverse effects of penicillins are allergic and nonallergic in nature.
PENICILLIN ALLERGY
Allergic reactions to penicillins are common while allergic fatalities are far less common. Allergy to penicillins ranges from 0.7% to 8% in various studies, with a 0.7% to 4% chance of an allergic reaction (average of 2%) during any given course of penicillin therapy.49,50 Most allergic manifestations are maculopapular or urticarial skin reactions.
Penicillin may be the most common cause of anaphylactic death in the United States, accounting for 75% of all cases and 400 to 800 annual deaths. These numbers may be low estimates, however. Penicillin-induced anaphylaxis is most common in adults 20 to 49 years old.49,50 Estimates of severe penicillin anaphylaxis range from 0.004% to 0.015% of individuals exposed and, from the point of view of number of exposures, possibly 1 in 1200 to 1 in 2500 penicillin exposures. The fatality rate from penicillin anaphylaxis by all routes of administration may be 1 in 60,000 patient courses (16 per 1 million),79,81 but data regarding penicillin allergy are limited.
Eventually, 1% to 10% of the general population exposed to therapeutic penicillin have an allergic reaction, with a higher positive history with increased age. Retrospective studies suggest that the incidence of allergy varies with the route of administration—oral (0.3%), intravenous (2.5%), and intramuscular (5%); the lower incidence by the oral route has been questioned because of limited data.79,80 With higher oral doses (3.5 g of amoxicillin), the allergy rate may approach that of intramuscular penicillin, indicating that the dose and the route may be a determining factor in penicillin allergy. Fatal anaphylactic reactions after oral penicillin are well documented.79,81
It is probable that an acute penicillin allergic reaction is less common in children and elderly patients, but fatal reactions may be more likely in elderly patients because of their compromised cardiopulmonary function. Whether certain individuals are predisposed to penicillin allergy remains unsettled. Risk factors for penicillin allergies include multiple allergies to other drugs, particularly other antibiotics (“multiple allergy syndrome”), or atopic disease (asthma, allergic rhinitis, nasal polyps). Several studies indicate a higher rate of penicillin allergy in individuals with a history of other drug allergies, whereas other studies indicate no such increased risk.79,81 It is possible that individuals with multiple drug allergies or atopy may have more severe penicillin allergic reactions. Allergy to the procaine component of procaine penicillin G has been detected.
In individuals with a positive history of penicillin allergy, 15% to 40% exhibit allergy on re-exposure to penicillin, and individuals with a positive history of penicillin allergy have a four to six times greater likelihood of a subsequent reaction than individuals with a negative history.79,81 Some patients may retain penicillin-specific IgE antibodies indefinitely, whereas most lose them over time. The serum half-life of penicillin IgE antibodies ranges from 10 to more than 1000 days; the risk of recurrent penicillin allergy is higher in individuals with antibodies with long half-lives or repeated penicillin exposures. Few data are available regarding whether the 60% to 85% not exhibiting allergy on re-exposure reacquire the IgE antibodies to penicillin and then have an allergic reaction to the drug on the next (third) exposure by resensitization. In a study of patients with a positive history of penicillin allergy (25 with urticaria/angioedema, 19 with anaphylaxis, 19 with pruritic skin rash) and a negative skin test for penicillin allergy, none had an allergic reaction to three successive 10-day courses of penicillin.91 The average length of time from the penicillin allergic reaction to rechallenge was 25 years (range 5 to 50 years), indicating that patients commonly lose their antibodies to penicillin. This study provides no information, however, on patients with a more recent history of penicillin allergy.
Because variable IgE antibody levels to penicillin are common, skin testing for penicillin allergy becomes problematic. The incidence of positive skin tests in individuals with a history of penicillin allergy ranges from 4% to 91% depending on the accuracy of the patient history, the haptens in the test solution, and the time elapsed between the allergic reaction and the skin test.68,70 It is possible that penicillin skin tests may be reliable only for 72 hours after the test is performed.79,81
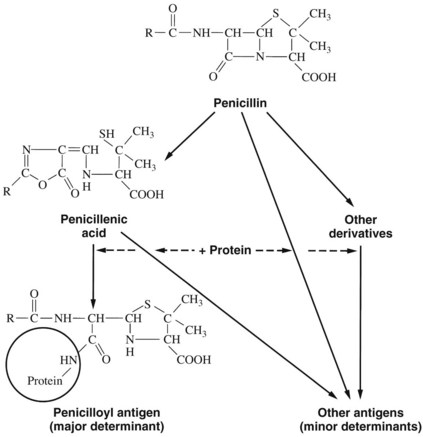
Penicillin skin testing can be considerably valuable in determining who might have a severe anaphylactic reaction. Approximately 95% of penicillin-allergic individuals form the penicilloyl-protein conjugate (the major antigenic determinant), and approximately 5% form the 6-aminopenicillanic acid and benzyl-penamaldic acid minor antigenic determinants (Figure 39-3).79,81 Penicillin skin tests with the major and minor antigenic determinants eliciting a negative skin test virtually eliminate the risk for a serious IgE-mediated reaction. A positive skin reaction to the minor determinant mixture indicates a high risk for anaphylaxis.
Allergic reactions to penicillins can also be classified according to their time of onset. Immediate IgE reactions begin within seconds to 1 hour after drug exposure and are the most life-threatening (it is an allergy truism that the more rapid the onset of the allergic reaction, the more serious the consequences). Accelerated reactions begin 1 to 72 hours after antigen exposure and usually manifest as urticaria or angioedema. Late reactions occur after 72 hours and are characterized by type II and type IV (eczema-like) Gell and Coombs reactions. Of all fatal anaphylactic reactions, 96% occur within the first 60 minutes after penicillin exposure.79,81
MULTIPLE ANTIBIOTIC ALLERGY SYNDROME
Most practitioners have encountered at least one patient with a history of allergy to multiple antibiotics (and probably other drugs as well). Whether such an antibiotic allergy syndrome exists is still undetermined, but may constitute 1% to 4% of all patients who have taken multiple antibiotics.66 Serious adverse drug reactions such as anaphylaxis with antibiotics are rare except for the β-lactams.88
Some difficulties with the health history are that (1) patients may confuse any adverse drug reaction with “allergy,” (2) rarely are any of these “allergies” confirmed by skin testing, and (3) often the patient’s history is nebulous.66 The management of such patients requires a detailed medical history, including (1) when during the treatment the reaction occurred, (2) what infectious disease was being treated, (3) what doses of antibiotics were taken and for how long, and, most important, (4) what were the signs and symptoms. The primary concern is to determine the signs and symptoms of an acute IgE-mediated allergic reaction—rash, angioedema, bronchospasm, and syncope—along with the time between drug ingestion or administration and onset of symptoms. If the onset of signs and symptoms occurred within 1 hour of ingestion or administration, it was likely an immediate allergic reaction. After taking a thorough history, a possible skin test (reliable only for β-lactams) is indicated and a specific management plan. Comorbid conditions that may increase the incidence of allergy in general are atopic disease (asthma, eczema), chronic urticaria, nonsteroidal anti-inflammatory drug (NSAID) intolerance, immunosuppression, human immunodeficiency virus (HIV) positivity, and a history of multiple antibiotic use.66
NONALLERGIC ADVERSE EFFECTS
Rare and reversible disorders reportedly associated with penicillins include acute pancreatitis, neutropenia, aseptic meningitis, hepatotoxicity, and increased prothrombin time/international normalized ratio (INR) in patients taking oral anticoagulants either through impaired platelet function or altered gastrointestinal microbial flora. Untoward bleeding may also occur in patients not taking coumarin anticoagulants and is dose-dependent, with a maximum effect 3 to 7 days after penicillin is begun, with a return to a normal bleeding time in 72 to 96 hours; this bleeding has been reported after a dental extraction.8 The mechanism is likely due to an altered adenosine 5′-diphosphate–mediated platelet aggregation response and is seen most commonly in patients with underlying chronic illnesses associated with hypoalbuminemia and uremia.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


