CHAPTER 35 Adrenal Corticosteroids
The adrenal gland is the source of a diverse group of hormones essential to metabolic control, regulation of water and electrolyte balance, and modulation of the body’s response to stress. The medullary portion of the gland secretes epinephrine and norepinephrine on sympathetic stimulation. These hormones and their physiologic effects and pharmacologic actions have been previously discussed (see Chapters 5 and 6). The adrenal cortex produces numerous substances derived from cholesterol and collectively known as corticosteroids. Certain corticosteroids and their synthetic analogues are used in medicine for replacement in adrenal insufficiency. They are used even more widely for an array of nonadrenal diseases, primarily for their notable ability to suppress the acute or chronic inflammation that accompanies injury and many diseases. When corticosteroids are administered in hyperphysiologic doses over long periods, as they often are in chronic inflammatory disorders, severe and even lethal toxicity can result. To understand the therapeutic applications and limitations of this group of drugs properly, the physiologic role of corticosteroids must be reviewed.
GENERAL PHYSIOLOGIC AND PHARMACOLOGIC ACTIONS
Using cholesterol as a substrate, the adrenal cortex synthesizes and secretes two types of steroid hormones—the 19-carbon androgens and the 21-carbon corticosteroids (Figure 35-1). The latter group can be considered derivatives of pregnane (Figure 35-2). The corticosteroids can be classified further on the basis of their major actions. Some of these compounds, such as hydrocortisone (the nonproprietary name for the natural hormone cortisol), have greater effects on carbohydrate metabolism, as measured by liver glycogen deposition, and are termed glucocorticoids. They also possess potent anti-inflammatory actions and are used therapeutically for this purpose. Other compounds, represented by aldosterone, are most active in enhancing Na+ retention and are referred to as mineralocorticoids. These corticosteroids do not have anti-inflammatory effects (Table 35-1). The structures of hydrocortisone and aldosterone are shown in Figure 35-2.

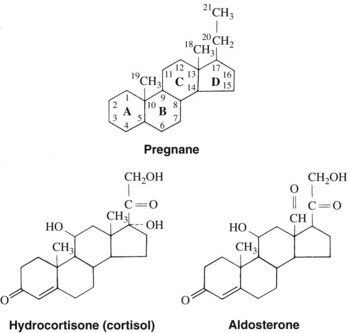
TABLE 35-1 Potencies of Commonly Used Corticosteroids (Relative to Hydrocortisone)
| LIVER GLYCOGEN DEPOSITION* | Na+ RETENTION | |
|---|---|---|
| 11-Desoxycorticosterone | 0 | 100 |
| Aldosterone | 0.1 | 3000 |
| Cortisone | 0.8 | 0.8 |
| Hydrocortisone | 1 | 1 |
| Prednisolone | 4 | 0.8 |
| Triamcinolone | 5 | 0 |
| Fludrocortisone | 10 | 3000 |
| Dexamethasone | 25 | 0 |
| Betamethasone | 25 | 0 |
* Generally paralleled by anti-inflammatory activity.
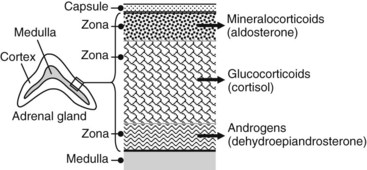
Production of corticosteroids and androgen is highly compartmentalized in the adrenal cortex (Figure 35-3). The mineralocorticoid aldosterone is produced by the outer layer cells (zona glomerulosa), cortisol and other glucocorticoids are produced by the middle layer (zona fasciculata), and the androgens are produced by the inner layer (zona reticularis) adjacent to the medulla. The corticosteroids are not stored to any extent in the adrenal gland but are continuously synthesized and secreted. The total daily production of the major glucocorticoid, cortisol, is normally 20 to 25 mg. There is a strong diurnal variation in this process; plasma concentrations of cortisol are severalfold higher at 8 a.m. than at 4 p.m.
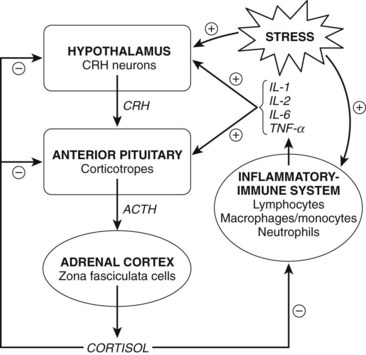
Production of all corticosteroids except aldosterone is directly regulated by the blood concentration of adrenocorticotropic hormone (ACTH) secreted by the anterior pituitary (adenohypophysis). Circulating corticosteroids act on the hypothalamus and adenohypophysis to suppress the release of ACTH, completing the control loop linking the pituitary and the adrenal cortex (Figure 35-4). By this negative feedback mechanism, the administration of large doses of corticosteroids can prevent the tropic influence of ACTH on the adrenal cortex, completely suppressing the adrenal production of corticosteroids. Aldosterone secretion is controlled primarily by a direct effect of angiotensin II on the adrenal cortex. Hyponatremia and hyperkalemia also favor the release of aldosterone. ACTH has only a minor stimulatory effect on the release of aldosterone.
Most of the diverse actions of corticosteroids seem to be achieved by regulating gene expression. As described in Chapter 32, glucocorticoids enter target cells and bind to cytosolic receptors. These receptors exist in a complex with several proteins, including heat shock protein and an immunophilin. Binding by the hormone or a synthetic analogue alters the conformation of the receptor, freeing it from the associated proteins. The hormone-receptor complex migrates to the nucleus and binds to the glucocorticoid-responsive elements on DNA of affected genes. The DNA-binding domain on the receptor is distinct from the drug-binding domain. Gene expression is regulated either negatively or positively to render the characteristic and complex glucocorticoid signature for modification of protein synthesis.14 The mechanism of action of mineralocorticoids has been less well studied, but it seems to be similarly based on regulation of transcription in renal and other target cells.
Consistent with the mechanism described, the major effects of corticosteroids are not manifested for several hours. Other, less apparent effects are more immediate, however. These likely occur through other receptor mechanisms involving the plasma membrane of target cells.7
The pharmacologic effects of glucocorticoids are largely exaggerations of the physiologic functions of the endogenous corticosteroids. These effects simulate the pathologic features of Cushing’s syndrome, a metabolic disorder resulting from an excess of corticosteroids, primarily cortisol. A review of these features and their pathophysiologic basis is helpful in understanding the pharmacologic and toxic actions of glucocorticoids.15
Carbohydrate and Protein Metabolism
Through several actions, glucocorticoids exert prominent anti-insulin effects. Glucocorticoids decrease the peripheral use of glucose by decreasing the cellular uptake of glucose. In the liver, glucocorticoids specifically stimulate glucose synthesis from amino acids (gluconeogenesis)8; concurrently, they mobilize amino acids by inhibiting protein synthesis in muscle, connective tissues, and skin (antianabolic effect). These actions are reflected in the parallel increases in blood glucose, liver glycogen, and urinary nitrogen excretion seen after the administration of glucocorticoids. As a consequence of these actions, prolonged high titers of glucocorticoids cause clinical manifestations of protein wasting: retardation of linear growth in children, wasting of the skin and increased capillary fragility resulting in ecchymoses, loss of muscle tissue leading to weakness (which may be extreme), and osteoporosis associated with enhanced bone resorption.
Anti-inflammatory Properties
Glucocorticoids are potent inhibitors of the inflammatory response. This anti-inflammatory activity is independent of the initiating stimulus and occurs at multiple points throughout the process (see Figure 35-4). The actions of glucocorticoids are due to many effects resulting from glucocorticoids binding to glucocorticoid receptors. The activated receptors bind to glucocorticoid response elements in the DNA, resulting in gene expression. In addition, activated glucocorticoid receptors can bind directly to transcription factors resulting in inhibition of inflammatory gene expression. Several resulting actions are especially prominent.
Glucocorticoids, by their action on gene expression, induce the production of numerous proteins with anti-inflammatory action, including annexin-1 (formerly lipocortin-1). Annexin-1 is a multifunctional protein that inhibits phospholipase A2 as one of its actions. The resulting decrease in the production of arachidonic acid leads to a reduction in the synthesis of prostaglandins and leukotrienes, important mediators of inflammation. (See Chapter 21 on nonsteroidal anti-inflammatory drugs [NSAIDs].) This effect is particularly important in macrophages, monocytes, endothelial cells, and fibroblasts. Glucocorticoids also suppress the synthesis of cyclooxygenase. The net result of these two actions is an inhibition of neutrophil, eosinophil, and monocyte chemotaxis (from inhibition of leukotriene B4 synthesis), inhibition of capillary permeability and bronchoconstriction (from decreased leukotrienes C4 and D4 and the prostaglandin PGF2α), and inhibition of the vascular and inflammatory responses to the prostaglandins PGI2 and PGE2.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


