CHAPTER 33 Drugs Acting on the Gastrointestinal Tract
Drugs that exert an effect on the gastrointestinal tract are among the most frequently used drugs. Digestive diseases are estimated to affect 60 to 70 million people in the United States each year with an annual direct cost of more than $85 billion.23 There is a high likelihood that a patient coming into the dental office may be on a regimen of one or more of these agents. Included in this group of drugs are anticholinergics, antihistamines, antacids, proton pump inhibitors (PPIs), antiemetics, laxatives, antidiarrheal or antispasmodic drugs, and gastrointestinal stimulants. Some of these drugs are available over-the-counter (OTC) without prescription and may be used at the discretion of the patient.
Many of the drugs discussed here are described in detail in other parts of the book. This chapter focuses on drugs used exclusively for their effect on the gastrointestinal tract and drugs with a wider spectrum of activity that have application to gastrointestinal disorders. Drugs that act on the gastrointestinal tract and are commonly used in dentistry to modify salivary gland activity or to reduce drug-induced nausea and vomiting are listed in Table 33-1.
TABLE 33-1 Drugs Useful in Dentistry That Affect the Gastrointestinal Tract
| THERAPEUTIC USE | DRUG | DOSE (mg)* |
|---|---|---|
| Sialagogue | Pilocarpine hydrochloride (Salagen) | 5† |
| Cevimeline hydrochloride (Evoxac) | 30† | |
| Antisialagogue | Atropine sulfate (Sal-Tropine) | 0.3-1.2 |
| Scopolamine hydrobromide (Scopace) | 0.4-0.8 | |
| Glycopyrrolate (Robinul) | 1-2 | |
| Propantheline bromide | 7.5-30 | |
| Antiemetic | Dimenhydrinate (Dramamine) | 50-100 |
| Meclizine hydrochloride (Antivert) | 25-50 | |
| Promethazine hydrochloride (Phenergan) | 25 |
GASTRIC HYPERACIDITY, GASTROESOPHAGEAL REFLUX DISEASE, AND PEPTIC ULCER DISEASE
Acid-peptic conditions such as heartburn (pyrosis), dyspepsia (indigestion), gastroesophageal reflux, and peptic ulcer disease (PUD) (gastric and duodenal) are often treated with drugs that either reduce intragastric acidity or promote gastrointestinal mucosal defense. In all these conditions, patient discomfort primarily results from the caustic effects of the gastric acid on the esophagus or from overcoming the gastrointestinal mucosal defense system or both. In the United States, heartburn has been reported to occur at least once a month by 44% of adults, at least weekly by 14%, and at least daily by 7%.12 Heartburn is a common term to describe a burning sensation that usually arises from the lower chest area (substernal) and moves upward toward the neck. It most commonly occurs within 2 hours after eating or when lying down or bending over. The symptoms are caused by the abnormal reflux of gastric contents or vapors retrograde into the esophagus. Heartburn that is frequent and persistent is the most common symptom of gastroesophageal reflux disease (GERD). GERD is one of the most prevalent digestive diseases among adults in the United States with more than 19 million cases annually.25 Symptoms arising from GERD, such as heartburn, are among the most common reasons for visits to primary care physicians.
PUD is a common malady affecting 10% to 15% of the population at some time in life. In a given year, nearly 15 million people in the United States have PUD.23 Although PUD is a painful condition that can seriously affect the quality of life, it is rarely fatal. Economically it is a major illness, with annual direct costs in the United States of greater than $3 billion, not including dollars lost in decreased wages and work productivity.25 Peptic ulcers are characterized by spontaneous healing and recurrence. The primary complication is hemorrhage, which may be life-threatening if undetected or ignored. Perforation of the gastrointestinal wall, which occurs much less frequently, accounts for most of the more than 4000 deaths from this disease each year in the United States.10
Throughout most of the twentieth century, therapy for PUD was directed at suppression of acid secretion or neutralization of secreted acid. This approach was based on the erroneous assumption that ulcers develop only because of increased gastric acid secretion. The primary causes of PUD are now known to be related to mucosal exposure to gastric acid and pepsin with a very strong association with Helicobacter pylori infection or the breakdown of normal mucosal defenses from the use of nonsteroidal anti-inflammatory drugs (NSAIDs).10 H. pylori infects more than half of the U.S. population older than 50 years and accounts for 80% of all stomach ulcers and greater than 90% of all duodenal ulcers.3 Because only a relatively small percentage of H. pylori–infected patients develop PUD in their lifetime, other factors must play a role in the development of this disease. Although H. pylori is found in saliva, the relationship between its presence in the mouth and infection in the stomach is unknown. The oral cavity may be a permanent reservoir for H. pylori, and a person-to-person route is the most probable mode of transmission.26
DRUGS USED TO REDUCE GASTRIC ACID AND TREAT PEPTIC ULCER DISEASE
Proton Pump Inhibitors
PPIs are drugs that irreversibly inhibit H+/K+-activated adenosine triphosphatase (H+,K+-ATPase, commonly called the proton pump) in the gastric parietal cell (Figure 33-1), the final common pathway for acid secretion. PPIs have become the drug class of choice for treating acid-related gastrointestinal diseases such as PUD and GERD. PPIs are among the most widely selling drugs because of their outstanding efficacy and safety. Currently, five members of the PPI class are available by prescription in the United States: esomeprazole, lansoprazole, omeprazole, pantoprazole, and rabeprazole (Table 33-2). Omeprazole is available OTC. When taken orally, all five agents effectively reduce basal and stimulated acid secretion considerably. They are longer lasting and substantially more potent than histamine H2-receptor antagonists in the short-term treatment of PUD and GERD and relief of heartburn.
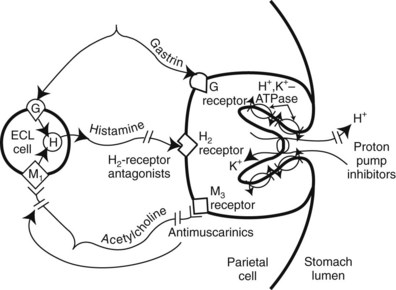

PPIs are administered as inactive prodrugs that accumulate selectively in the acid environment of the secretory canaliculus of the gastric parietal cell. The PPI is rapidly protonated and converted to the active form of the drug. Because PPIs bind covalently to active proton pumps, synthesis of new pumps or activation of resting pumps is required to restore activity. This irreversible inhibition of the pump explains why the duration of action of this class extends beyond the elimination half-life of 0.5 to 2 hours (see Table 33-2). PPIs are best taken on an empty stomach (food can decrease bioavailability up to 50%) once daily 1 hour before a meal so that the peak serum concentration coincides with the maximum activation of the proton pumps.
The most common adverse effects reported with PPIs are headache, diarrhea, and nausea, but the frequency is only slightly greater than placebo. Long-term use of PPIs may cause a slight increase in serum gastrin. This information led to concerns regarding gastrin-induced neoplasms that have been reported in animal models. PPIs have been available for more than 20 years and, to date, none have been associated with an increased risk of gastric cancers in patients receiving long-term therapy. Of more recent concern are reports that PPIs, particularly at high doses, are associated with an increased risk of hip fracture by interfering with Ca++ absorption through induction of hypochlorhydria31 and with an increased risk to develop community-acquired Clostridium difficile–associated disease (CDAD).22
All PPIs increase gastric pH and may alter the absorption of drugs that are weak bases or acids or formulated as pH-dependent, controlled-release products. Absorption of aspirin, digoxin, and midazolam may be increased, and ketoconazole absorption may be decreased when administered with a PPI. The clinical significance of the alterations is unclear. PPIs can also alter the hepatic metabolism of other medications. All PPIs are metabolized to varying degrees by hepatic P450 cytochromes, including CYP2C19 and CYP3A4, and may interfere with the medications metabolized by these same enzymes. Omeprazole has been shown to progressively inhibit CYP2C19 activity with repeated administration and may inhibit the metabolism of diazepam, warfarin (Coumadin), and phenytoin.24 Despite these concerns, few clinically significant drug interactions have been reported given the enormous popularity of PPIs.
H2 Receptor Antihistamines
Histamine is one of the primary mediators of gastric acid secretion, along with acetylcholine and gastrin. The final common pathway is through the proton pump (see Figure 33-1). As discussed in Chapter 22, H2 receptors are located on the membranes of acid-secreting parietal cells of the stomach. H2 receptor antihistamines (commonly called H2 blockers) are reversible, competitive antagonists of histamine at the H2 receptors. The duration and the degree of acid suppression are dose-dependent. These are highly selective agents in that they do not affect the H1 receptors and are not anticholinergic. Cimetidine, the first of these drugs to be used widely, revolutionized the treatment of duodenal ulcers. With the recognition of the role of H. pylori in PUD and the introduction of PPIs, the use of H2 antagonists has markedly declined.
A usual single dose of any of the H2 antagonists currently available for prescription or nonprescription use in the United States, including cimetidine, famotidine, nizatidine, and ranitidine (Table 33-3), inhibits 60% to 70% of total 24-hour acid secretion. These agents are particularly effective in inhibiting nocturnal acid secretion, which is stimulated more by histamine. Food-induced gastric acid secretion is stimulated more by gastrin and acetylcholine and is less inhibited by the H2 blockers.

H2 blockers are commonly administered orally. The antisecretory activity usually begins within 1 hour of administration and persists for 6 to 12 hours. They have an oral bioavailability of 40% to greater than 90%, achieve peak plasma concentrations in 0.5 to 3 hours, and are eliminated with a terminal half-life of 1.5 to 3 hours (see Table 33-3). The drugs undergo partial metabolism in the liver; the remainder of the parent drug is eliminated unchanged by the kidney. The duration of effectiveness varies with the drug, dose, and medical condition being treated, ranging from 4 hours for a low dose of cimetidine for hypersecretory disorders to 24 hours for all these agents when used to treat duodenal and gastric ulcers.
Comparative studies of H2 blockers show that the four drugs in this class are essentially equal in clinical effectiveness regarding ulcer treatment even though they express varying potencies in their ability to block pentagastrin-stimulated gastric acid secretion in the research laboratory. Cimetidine seems unique among H2 blockers in exerting biologic effects that are unrelated to gastric H2 occupancy. Cimetidine therapy, particularly when prolonged and at high doses, can cause antiandrogenic effects. These reversible effects result from the ability of cimetidine to compete with dihydrotestosterone at androgen-binding sites and to inhibit the CYP metabolism of estradiol.13 Men treated with high doses of cimetidine for long periods may experience impotence and development of gynecomastia, whereas women may develop galactorrhea. Substitution of ranitidine for cimetidine reverses these effects; no antiandrogenic effects have been reported after therapeutic doses of famotidine or nizatidine.
Of importance to the dentist is the ability of cimetidine to decrease the hepatic oxidative biotransformation of many other drugs, including lidocaine and diazepam. Cimetidine and ranitidine are ligands for multiple CYP enzymes (see Table 2-3), with cimetidine exhibiting a much higher affinity and inhibiting hepatic microsomal enzyme activity to a much greater extent. The clinical use of ranitidine, famotidine, and nizatidine does not seem to have a significant effect on the metabolism and elimination of other drugs.
Antibiotics
The evidence that PUD (and gastritis and possibly gastric adenocarcinoma) is directly linked to infection by the gram-negative organism H. pylori is now well established. Cultures taken from biopsy material are positive for H. pylori in approximately 95% of duodenal ulcer specimens and 75% of biopsy specimens taken from gastric ulcers compared with a roughly 25% incidence in asymptomatic control subjects.5
These findings have led to the routine use of antibiotic therapy for the eradication of gastric and duodenal ulcers. Significant reductions in clinical symptoms and histologic evidence of ulcers have been achieved. The current cornerstone of therapy for H. pylori–associated peptic ulcers involves a triple regimen of a PPI (e.g., lansoprazole) with dual antibiotics clarithromycin and amoxicillin. This treatment regimen results in eradication of the organism in greater than 80% of patients,9 although the success rate has been declining because of increasing clarithromycin resistance.6 PPIs not only add antisecretory properties, but may also enhance healing through direct anti–H. pylori properties. Other therapeutic approaches include adding bismuth subsalicylate to the regimen (quadruple therapy) or substituting a different antibiotic such as levofloxacin or metronidazole.9 For patients with NSAID-induced PUD, rapid healing is often initiated with the use of a PPI and discontinuation of the NSAID. Future studies are required to determine the exact interaction between bacterial infection and other prognostic factors (e.g., smoking, alcohol, NSAIDs) implicated in ulcer formation.
Gastric Antacids
Calcium carbonate
Calcium carbonate produces a potent and prolonged neutralization of HCl forming CO2 and CaCl2. Approximately 90% of the ingested Ca++ forms insoluble salts in the gut and is excreted in the feces. The remaining Ca++ is absorbed into the systemic circulation. Extensive use of Ca++-containing antacids may cause or exacerbate hypercalcemia, which is characterized by neurologic symptoms and reduced renal function. This effect is rare in healthy patients with normal renal function. Ca++-containing antacids are associated with acid rebound and increased serum gastrin concentrations. These effects have not been shown to delay ulcer healing and may be caused by a direct effect of Ca++ on the gastric mucosa.25 Calcium carbonate has a chalky taste and may produce constipation, which reduces its desirability as an antacid. Because some Ca++ is absorbed, Ca++-containing antacids may be marketed as a source of dietary Ca++.
Sucralfate
Although sucralfate has multiple actions, it possesses no meaningful antacid properties. A key element in the acute gastroprotective actions of sucralfate is its ability to maintain mucosal vascular integrity and blood flow. It enhances bicarbonate and mucus secretion, increases mucosal hydrophobicity, and induces an increase in mucosal concentration of prostaglandin—all factors considered important in tissue healing. An increase in local fibroblast growth factors and possibly other growth factors has also been proposed to explain the powerful ulcer-healing actions of sucralfate, which occur independently of a decreased gastric acid concentration in the stomach and duodenum.7
Because it is minimally absorbed from the gastrointestinal tract, sucralfate is considered a remarkably safe agent. For this reason, sucralfate is a first-choice therapy in the management of acid-related diseases during pregnancy.8 It requires an acid pH to be activated and so should not be administered concomitantly with antacids, H2 antagonists, or PPIs. The most common side effect is constipation (15%). Other reactions include dry mouth, nausea, vomiting, headache, and rashes.
Sucralfate may reduce the absorption of many other drugs, including the fluoroquinolone and tetracycline antibiotics. The use of a topical sucralfate suspension has also been advocated in the prevention or treatment of stomatitis caused by chemotherapy or radiation, despite studies that showed no substantial benefits from this drug in inhibiting radiation-induced esophagitis.21
Antimuscarinic Drugs
The use of antimuscarinic drugs (muscarinic receptor antagonists) for the treatment of PUD declined dramatically after the introduction of the H2 blocker cimetidine. As discussed in Chapter 9, antimuscarinic agents (e.g., atropine) are not selective inhibitors of gastric acid secretion, and therapeutic benefits for the treatment of gastrointestinal disease accrue only at doses that cause sufficient side effects to impair patient compliance. Antimuscarinic drugs with a higher relative affinity for gastric M1 muscarinic receptors have been developed, however. Pirenzepine and telenzepine, selective M1-receptor antagonists, are currently available in other countries for the treatment of PUD, but they are still investigational in the United States. Pirenzepine and telenzepine block gastric acid secretion more selectively because the M1 receptor is not the major muscarinic receptor in most smooth muscle, cardiac muscle, or salivary glands. In those tissues, M2 and M3 muscarinic receptors predominate. Pirenzepine and telenzepine have a low incidence of side effects because of their selective inhibition of gastric acid secretion; this may make them a valuable addition to current agents used in the treatment of PUD.
Prostaglandins
Misoprostol, a synthetic prostaglandin E1 analogue, is the best studied of the prostaglandin derivatives. Although the prostaglandins are crucial in creating the alkaline mucus layer that provides cytoprotective effects on the gastroduodenal mucosa, the ulcer-healing effect of misoprostol and other prostaglandin analogues seems to be caused mainly by the inhibition of acid secretion.13 These agents interact with a basolateral receptor of the parietal cell that causes the inhibition of adenylyl cyclase. This inhibition results in reduced production of cyclic adenosine 3′,5′-monophosphate, the major second messenger for histamine-induced acid secretion. Misoprostol is approved for prevention of NSAID-induced ulcers in high-risk patients, although PPIs may be as effective and better tolerated. The most common side effects are abdominal pain (7% to 20%) and diarrhea (13% to 40%); both are dose-related. Misoprostol stimulates contraction of the uterus, which contraindicates its use during pregnancy or in women of childbearing potential. This property makes it effective, however, in women undergoing elective termination of pregnancy by facilitating expulsion of the uterine contents.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


