CHAPTER 32 Drugs Acting on the Respiratory System
Worldwide interest and concern continue to increase as a result of the escalating morbidity of inflammatory lung disease. According to the Centers for Disease Control and Prevention, the prevalence of asthma among U.S. children increased from 3.8% in 1980 to 5.8% in 2003.27 Globally, it is estimated that 300 million people have asthma.32 The World Health Organization in the Global Burden of Disease Study ranked chronic obstructive pulmonary disease (COPD) as the 12th leading contributor to the burden of disease in 1990; by 2020, it is estimated that COPD will be ranked 5th.45 COPD is currently the fourth leading cause of death in the United States.55
PATHOPHYSIOLOGY OF ASTHMA
Asthma is a chronic inflammatory disease of the airways associated with acute symptoms, exacerbations, and airway remodeling.14 The acute symptoms, bronchospasm and wheezing, can be reversed by bronchodilators. Exacerbations and airway remodeling are caused by chronic inflammation. Exacerbations can be controlled with anti-inflammatory drugs. There is no defined treatment for airway remodeling. Airway obstruction follows an acute bronchoconstrictor response to the spasmogenic stimulus, peaking within 10 to 20 minutes.15 This early-phase reaction is characterized by the release of IgE and the activation of cells bearing allergen-specific IgE receptors, especially airway mast cells. The activated cells produce proinflammatory mediators such as histamine, eicosanoids, and reactive oxygen species (ROS).
PATHOPHYSIOLOGY OF CHRONIC OBSTRUCTIVE PULMONARY DISEASE
COPD is a progressive condition characterized by irreversible airflow limitation.41 The major etiologic factor is cigarette smoking. Cigarette smoke is a complex mixture of approximately 4700 compounds, including high concentrations of free radicals and other oxidants. Inflammation of the lungs as a reaction to cigarette smoke is characteristic of the disease and is thought to produce lung injury. Symptoms of COPD include chronic cough with sputum production and breathlessness on exertion. There is also a systemic component to the disease involving various organs, including skeletal muscle, the central nervous system (CNS), and the cardiovascular system. Systemic inflammation intensifies with progression of COPD and has been linked to cardiovascular events, muscle wasting, and colon cancer.
DRUGS USED TO TREAT ASTHMA
Corticosteroids
Mechanism of action
Corticosteroids diffuse readily across plasma membranes to enter cells where they encounter the glucocorticoid receptor, a cytosolic protein. When the steroid binds the receptor, the receptor undergoes a conformational change.8 Chaperone proteins dissociate from the receptor, and nuclear receptor localization regions on the receptor are exposed. The receptor-steroid complex dimerizes and is transported to the nucleus, where it binds to DNA in promoter sequences termed glucocorticoid response elements (GREs). Repression of genes activated by inflammatory diseases is the predominate effect of corticosteroids. These genes encode cytokines; chemokines; adhesion molecules; and inflammatory enzymes, proteins, and receptors. Repression of these genes by corticosteroids occurs at concentrations easily achieved clinical practice. There are no recognizable GRE sites within these genes, however. This response seems to be a result of corticosteroid effects on chromatin remodeling.6
Chronic inflammation involves activation of the proinflammatory transcription cofactors nuclear factor-κB (NF-κB) and activating protein-1 (AP-1). NF-κB and AP-1 bind to large coactivator molecules, such as cyclic adenosine 3′,5′-monophosphate (cAMP) response element–binding protein, which possess intrinsic histone acetyltransferase activity. Acetylation of core histones on specific lysine residues reduces their charge and changes the electrostatic attraction between the histone protein and DNA; this allows the histone-DNA complex to undergo a conformational change from the closed structure to the activated open form. DNA unwinds and is accessible to RNA polymerases, which initiate transcription of inflammatory genes. Histone deacetylases (HDACs) act as corepressors along with other proteins, such as nuclear receptor corepressor, to silence gene expression. Deacetylation of histone proteins on lysine residues returns the DNA to the closed basal state, countering the activity of the histone acetyltransferases.1
Corticosteroids have multiple effects on chromatin remodeling proteins. Anti-inflammatory genes are activated by selective acetylation of histone H4 by coactivator molecules, such as steroid receptor coactivator-1 (SRC-1) and glucocorticoid receptor interacting protein-1 (GRIP-1). Corticosteroids also repress gene activity by reversal of histone acetylation of proinflammatory sequences; this can occur in two pathways. First, activated glucocorticoid receptor can bind directly to cAMP response element–binding protein and other coactivators and inhibit their histone acetyltransferase activity. Second, and more important, activated glucocorticoid receptor can recruit HDACs to the transcriptional complex, which results in deacetylation of histones associated with inflammatory gene expression.2
Steroids can also reduce chronic inflammation by transactivation.8 Transactivation requires higher concentrations of steroids, however, than those concentrations required for chromatin remodeling. Transactivation involves the monomeric glucocorticoid receptor complex, which can interact directly with AP-1 and NF-κB through protein-protein interactions. This interaction decreases their transcription factor–mediated proinflammatory actions. Corticosteroids also increase the expression of two proteins that affect these inflammatory signal transduction pathways: glucocorticoid-induced leucine zipper protein (GILZ) and mitogen-activated protein kinase phosphatase-1 (MKP-1). GILZ inhibits AP-1 and NF-κB. MKP-1 inhibits p38 mitogen-activated protein kinase, which is activated by AP-1.
Pharmacologic effects
The pharmacologic effects of steroids on inflammatory lung diseases indicate that they are very effective at controlling symptoms of the disease, but they do not cure the disease. When the steroid dose is reduced, or the steroid is discontinued, the symptoms eventually return. In asthma, corticosteroids reduce airway inflammation and hyperresponsiveness, improve lung function, and decrease the incidence and severity of acute asthma exacerbations. Treatment of COPD with corticosteroids is controversial.42 Experimental data indicate that the inflammatory processes in COPD may be resistant to the effects of corticosteroids because of oxidation and nitrosylation of proteins involved in chromatin remodeling by the oxidants found in cigarette smoke. Clinical data show inhaled corticosteroids improve the health of patients with COPD, however.50 The pharmacologic effects seen with corticosteroids in COPD are largely the same as those seen with asthma, but they also reduce systemic inflammation and reduce the rate of decline of health in patients with COPD.
Efficacy and safety
The optimal route of administration of corticosteroids for inflammatory lung disease is inhalation. Inhalation delivers the drug directly to the lungs. The corticosteroid acts locally, which minimizes the systemic adverse effects encountered with oral or parenteral administration. Receptor-binding affinity is the only pharmacodynamic parameter that differs among the corticosteroids that are available for inhalation.24 High binding affinity can be considered a desirable trait providing for better efficacy in the lung. High binding affinity can also be detrimental because it can lead to increased systemic adverse effects.
The differences between the inhaled products depend on factors such as formulation, pulmonary and systemic bioavailability, and excretion.23 Characteristics that enhance the efficacy of inhaled corticosteroids include small particle size and long pulmonary residence time. Solution aerosols formulated with hydrofluoroalkane, such as ciclesonide and beclomethasone dipropionate, have a small mass median aerodynamic diameter (MMAD) <2 µm. A small MMAD can have effects on efficacy because the smallest airways have an internal perimeter ≤2 µm. MMAD can also have an impact on local adverse effects because larger particles are more likely to be deposited in the oropharyngeal cavity.
Pulmonary residence time depends on two factors, lipophilicity and lipid conjugation. Lipophilic side chains on the D-ring of the steroid portion of the drug (see Figure 35-2) slow the dissolution in aqueous bronchial fluid. Lipophilic groups also aid the passage of the drug through the phospholipid bilayer of cell membranes to reach the receptors in the cell. Lipid conjugation to fatty acids in the pulmonary cells occurs via a reversible ester bond. Esterification provides a slow-release reservoir of the corticosteroid, prolonging residence time. This prolonged residence time allows a once-a-day dosing schedule for budesonide and ciclesonide.
Adverse effects
The most common adverse effects of inhaled corticosteroids are oropharyngeal candidiasis and dysphonia.36 Although they can be easily managed, these localized adverse effects diminish compliance leading to uncontrolled disease and reduced quality of life. Less common adverse effects of inhaled corticosteroids are systemic in nature, including adrenal suppression, bone loss, skin thinning, metabolic changes, behavioral abnormalities, weight gain, and decreased linear growth in children. The concentration of the corticosteroid in the blood is a combination of the portion that is swallowed and the portion that is delivered to and absorbed from the lungs.
The safety of inhaled corticosteroids depends on factors such as activation in the lungs, low oral bioavailability, high protein binding, and rapid systemic clearance. Although most corticosteroids are inhaled in their pharmacologically active form, beclomethasone dipropionate undergoes activation in the human lung.30 Beclomethasone dipropionate is metabolized to beclomethasone 17-monopropionate, which has a relative receptor affinity about 30 times greater than the parent compound. Metabolism in human plasma is quite different from metabolism in human lung. Human lung metabolism involves a simple hydrolysis reaction to the active form. Metabolism in human plasma involves hydrolysis, transesterification, and loss of hydrogen chloride. Metabolism in human plasma results in beclomethasone 21-monopropionate, which has no binding affinity for the glucocorticoid receptor. This differential metabolism in lung and plasma leads to fewer systemic effects of beclomethasone.
β2-Adrenergic Receptor Agonists
β2-adrenergic receptor agonists are structurally modified catecholamines. The useful structural modifications result in increased β2 receptor selectivity, enhanced oral activity, and an extended duration of action after inhalation.12 Susceptibility to degradation by catechol-O-methyltransferase (COMT) and reuptake mechanisms in neurons are decreased by structural modifications. There are two types of β2 adrenergic receptor agonists. The short-acting drugs have bronchodilatory effects that last 4 to 6 hours and are exemplified by albuterol. The long-acting drugs have bronchodilatory effects that last 12 hours and bronchoprotective effects lasting 6 to 12 hours.
Short-acting and long-acting drugs were designed along the same structure-activity principles. Albuterol and salmeterol have a nearly identical active moiety, (Figure 32-1). Salmeterol has an extended aliphatic side chain, however, which was designed to anchor the drug in or near the receptor. There are currently two long-acting β2 agonists on the market: salmeterol and formoterol.3 Formoterol is a formanilide-substituted phenylethanolamine. Formoterol has a higher intrinsic efficacy and a faster onset of action than salmeterol.
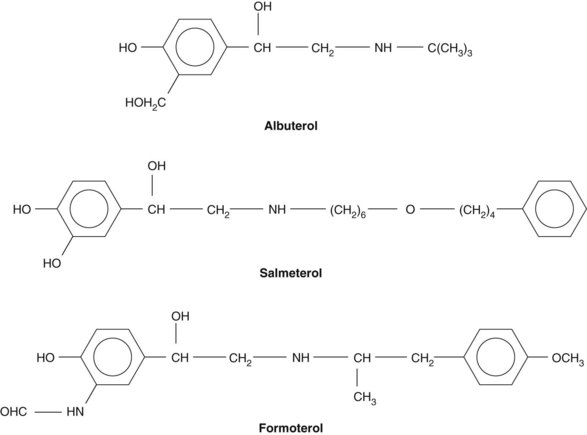
Two theories have been proposed to explain the increased duration of action of salmeterol and formoterol. Using site-directed mutagenesis of the β2 receptor, Green and coworkers34 proposed that the aliphatic side chain of salmeterol binds in a second locus of the receptor that is separate from the active site, termed the exosite. These investigators used β receptor antagonists to determine activity kinetics. An exosite would explain how activity is so quickly restored after washout with the β receptor antagonist. In the second theory, termed the diffusion microkinetic theory, which applies to salmeterol and formoterol, the high membrane partition affinity of the two drugs creates a microdepot of the drug near the receptor.3 The exact nature of the sustained duration of action is likely to relate to aspects of the clinical pharmacology and tolerability of salmeterol and formoterol.
Mechanism of action
The β2-adrenergic receptor is a member of the seven-transmembrane receptor superfamily of G protein–coupled receptors.2 The ligand-binding pocket open to the extracellular space is formed when the seven α helices of the receptor cluster together in a ring. When a receptor is moved to its active state by binding of ligand, its associated G protein dissociates into a Gα subunit and a β/γ dimer. As illustrated in Figure 5-7, the Gαs subunit activates adenylyl cyclase, which increases the concentration of cAMP in the cell. cAMP activates protein kinase A. Protein kinase A phosphorylates protein substrates that control Ca++ availability in the cell. With decreased Ca++ availability, the myosin light chain is ineffective in sustaining active tone in airway smooth muscle. The tissue relaxes passively.
The mechanism previously described is the traditional cAMP/cAMP-dependent protein kinase A cascade. Studies since the 1990s suggest that there are other mechanisms involved with β-adrenergic signaling in the airways.31 In addition to Gαs, the β2-adrenergic receptor can also couple to Gαi and inhibit adenylyl cyclase. β2 Receptors are found on proinflammatory and immune cells, including mast cells, macrophages, neutrophils, lymphocytes, eosinophils, epithelial and endothelial cells, and types 1 and 2 alveolar cells. Inhibition of adenylyl cyclase may be involved in decreasing the reactivity of these proinflammatory cells.
Adverse effects
The most serious adverse effect of β2 agonist use is an increase in sensitivity of the smooth muscle in the lungs to noxious stimuli. Paradoxically, an increased incidence of asthma exacerbations and morbidity and mortality is seen after long-term use of β2 agonists44; this is attributable to the loss of bronchoprotection and bronchodilation effects of the drugs. This effect is called tolerance, a state of refractoriness that occurs after prolonged exposure to an agonist. The mechanistic basis for tolerance involving β-adrenergic receptors is currently unclear. Factors that can contribute to tolerance include receptor downregulation, due to receptor internalization, and receptor desensitization (see Figure 1-12) which can be due in part to receptor uncoupling from adenylyl cyclase.15 It is not the use of intermittent short-acting β2 agonists, such as albuterol, that leads to tolerance, but rather the continued use of long-acting β2 agonists.
The U.S. Food and Drug Administration (FDA) has required manufacturers of long-acting β2 agonists to place a “black box” warning on the physician information for salmeterol and formoterol. Data for the advisory labeling came from the Salmeterol Multi-Center Asthma Research Trial (SMART),18 which examined the safety of salmeterol when added to usual asthma therapy. Study subjects who were on salmeterol showed a small but significant increase in asthma-related death compared with subjects receiving placebo.
With the exception of levalbuterol, all β2 agonists are racemic mixtures of R-enantiomers and S-enantiomers.39 Although the R-enantiomer of albuterol, levalbuterol, is 100 times more potent at the receptor, the S-enantiomer of albuterol is metabolized much more slowly and is retained in the lung. Preclinical data indicate that S-albuterol is associated with proinflammatory activity that can lead to bronchial hyperresponsiveness. These activities include an increase in intracellular Ca++, an increase in inflammatory stimuli, an increase in eosinophil activation and recruitment, and an increase in mucin production. These proinflammatory activities have led to speculation that the S-enantiomer of salmeterol is responsible for the loss of effect seen with long-term use of this drug. Formoterol has two chiral centers, and there is evidence to suggest that S-formoterol accumulates in bronchial tissue relative to the R-enantiomer because of different rates of metabolism. Many new long-acting β2 agonists in clinical development are pure active enantiomers.
Specific agents
Albuterol
Albuterol, the most commonly prescribed β2-adrenergic bronchodilator, is available in tablet form, as a sustained-release tablet, and as a metered-dose pressurized aerosol. Pulmonary β2 specificity is greater with aerosol delivery than it is with an oral dose.40 Adverse reactions to albuterol are similar to adverse reactions of other β2-adrenergic receptor agonists.
Salmeterol
Incorporating a long-acting β2 agonist into the therapeutic regimen has several advantages. Clinical trials have shown salmeterol to be more efficacious than albuterol in (1) reducing variation in diurnal peak expiratory flow rates; (2) decreasing nocturnal symptoms, asthma exacerbations, and the need for rescue medications; and (3) increasing overall lung function.20,49 The combination of salmeterol with an inhaled corticosteroid has been shown to be beneficial. Several studies have indicated that the administration of salmeterol with a lower dose of corticosteroid results in improved pulmonary function compared with a larger dose of corticosteroid administered alone.29,61 In addition to the improved physiologic response, the administration of salmeterol decreases the potential of systemic effects of corticosteroids.
Terbutaline
Terbutaline, a congener of metaproterenol, is of clinical interest because of its long duration of action (3 to 6 hours), its availability in an injectable form, and a side-effect profile equivalent to metaproterenol.4 For patients with spontaneous asthma, terbutaline is reported to provide greater protection against bronchoconstriction than an equivalent dose of metaproterenol. Terbutaline from the aerosol metered-dose inhaler stimulates bronchial β2 receptors more selectively than oral or parenteral forms of the drug. Tolerance to the aerosol does not seem to develop with continued use over several months. The most common side effect of therapeutic doses is a slight tremor, which may be particularly noticeable in elderly patients. Other side effects, typically seen with nonselective β blockers, are infrequent. Terbutaline is the only drug used as a bronchodilator that has good safety data supporting its use during pregnancy.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


