CHAPTER 31 Procoagulant, Anticoagulant, and Thrombolytic Drugs
HEMOSTASIS
Platelet Adhesion, Activation, and Aggregation
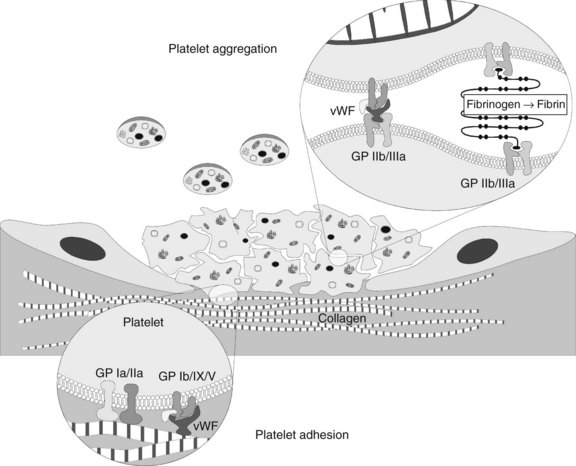
Adhesion
The presence of these proteins, particularly vWF, stimulates a “catch and grab” response in the platelets, causing them to leave the laminar flow of the blood and adhere to the injured area. Platelets have a high density of surface receptors that respond to these proteins, and they undergo an extremely rapid localization to the site of injury, beginning the formation of a thrombus. Two main receptors are involved in adhesion: the glycoprotein (GP) Ia/IIa heterodimer, which binds to collagen directly but weakly, and the GP Ib/IX/V heterotrimer, which binds with high shear strength to connective tissue vWF associated with the collagen surface (Figure 31-1).1 The GP Ib/IX/V–vWF linkage is more of a “tethering” of the platelet to the substrate; later, the adhesion is firmed up by GP IIb/IIIa activation. If vessels without a muscular sheath are severed, the immediate hemostatic action of platelet aggregation is especially important. The true significance of platelets in hemostasis is most evident in the management of patients with thrombocytopenia.
Activation
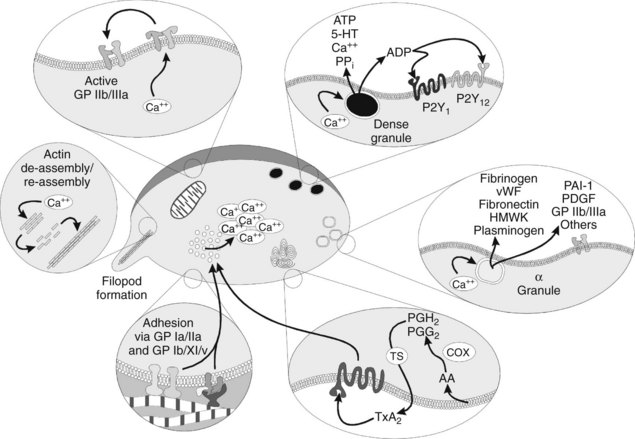
Activation of platelets is a crucial step in forming a proper thrombus. Activation can occur from various agonists, some of which are strong and some of which are weak. Examples include thrombin, adenosine diphosphate (ADP), thromboxane A2 (TXA2), 5-hydroxytryptamine (serotonin), epinephrine, vasopressin, fibrinogen, immune complexes, plasmin, and platelet-activating factor. Most plasma-derived agonists exert their effect by numerous G protein–linked membrane receptors. The strongest agonist for platelet activation is binding of vWF to the GP Ib/IX/V heterotrimeric receptors.14 When one of these receptors is bound by its specific agonist, an intraplatelet protein cascade begins that ultimately causes activation of Ca++ transporters and movement of Ca++ from stores in the platelet’s dense tubular system to the general intracellular matrix.26 The intracellular increase in Ca++ causes several other changes.
Platelets in the resting state have internal cytoskeletal actin that provides them with a smooth shape; as Ca++ increases, the actin is initially fragmented into smaller subunits, transforming the normal discoid shape of the platelet to a spherical conformation. These smaller actin subunits are rapidly reassembled into very-long-chain actin monomers, which cause the platelet to sprout filopods. The filopods are important in ultimate clot retraction. Meanwhile, as the filopods are developing, the increasing intracellular Ca++ concentrations act on cytoplasmic vesicles known as α and dense (or δ) granules (Figure 31-2), prompting them to rise to the cell surface and degranulate. The dense granules release ADP, adenosine triphosphate (ATP), the vasoconstrictor 5-hydroxytryptamine, Ca++, and inorganic pyrophosphate.18 The α granules contain numerous proteins involved in coagulation, adhesion, cellular mitogenicity, protease inhibition, and other functions (Box 31-1). Major proteins released include fibrinogen, coagulation factors, vWF, fibronectin, high-molecular-weight kininogen, plasminogen, plasminogen activator inhibitor-1 (PAI-1), platelet-derived growth factor, additional GP IIb/IIIa, and thrombospondin.18
Aggregation
As the α and dense granule contents are released extracellularly, nearby platelets become activated. The ligand proteins bind to the surface-associated GP IIb/IIIa of these adjacent platelets, forming bridges. At low shear rates, fibronectin and fibrinogen (stabilized by thrombospondin, another GP from the α granules) serve as the main adhesive proteins, whereas vWF is necessary for proper adhesion in areas of high shear. Microvascular video imaging studies show that thrombus formation initially is inefficient. Platelets bind quickly, but a significant percentage of them break free and float away. As a result, thrombus formation is much slower than would be the case if all the platelets that physically aggregate remained bound.1
Perhaps the most remarkable effect of platelet activation is the procoagulant activity the platelets impart. In the normally resting platelet, the plasma membrane has negatively charged phospholipids, including phosphatidylserine, sequestered almost exclusively on the inner surface by processes that are not fully understood. When activating ligands bind to the platelet, the resultant increase in intracellular Ca++ causes a membrane enzyme termed scramblase to evert the phosphatidylserine to the outer surface, while simultaneously prompting the membrane to form small evaginated microvesicles. Factors Va and VIIIa (discussed subsequently) bind to the phosphatidylserine moieties and recruit factors Xa and IXa. The interaction of these complexes in toto accelerates the conversion of prothrombin to thrombin by a factor of 2.4 × 106. In addition, the binding of activated coagulation factors to the platelets seems to protect the factors from plasma inhibitors, while directing the bulk of the coagulation cascade to the site of injury. The α granules contain factors V and IX; factor V is apparently complexed with multimerin, a carrier protein (see Box 31-1).
As the thrombin is generated, it activates other platelets by stimulating G protein–linked receptors. The thrombin receptors seem to be unique “suicide” receptors, requiring proteolytic cleavage to transmit an activating signal. Thrombin is a serine protease, and it acts on the receptors by cleaving the protein at a serine residue near the amino terminus. The new amino terminus acts as a “tethered ligand” to double back and stimulate the transmembrane protein to activate—hence this receptor has been named a protease-activated receptor (PAR). There are four such thrombin receptors, PAR-1 through PAR-4; only PAR-1 and PAR-4 are expressed by human platelets.23 Thrombin-induced activation seems to upregulate GP IIb/IIIa activation while downregulating GP Ib/IX/V activity. The platelets apparently are converted from a mainly adhesive role to an aggregate role when thrombin is present.
Two other important activities of platelets warrant mention. First, the α granules contain P-selectin, a membrane protein that helps recruit and tether neutrophils and monocytes into the local area. This activity is believed to be crucial for generating a local inflammatory response at the site of injury, while promoting yet limiting thrombosis.35 Second, platelets are also essential in clot retraction, an event that facilitates wound healing by bringing the severed ends of small blood vessels into closer apposition. Clot retraction, or syneresis, occurs when the filopodia expressed by platelets during activation attach to fibrin strands and contract. A number of actin-binding proteins are present in platelets.4 On activation, phosphorylated myosin monomers polymerize into filaments next to the long-chain actin filaments, which slide past one another to generate a contractile force in the presence of ATP.
Coagulation Cascade
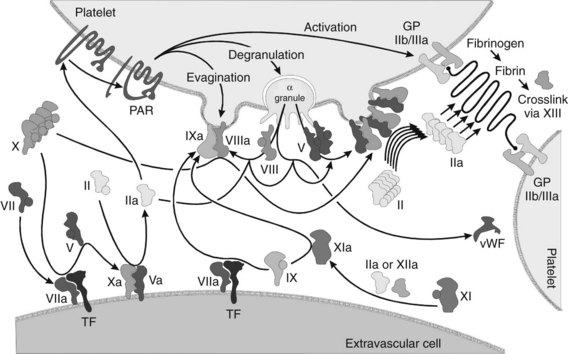
Although it is possible to separate the numerous events of hemostasis (e.g., platelet aggregation, formation of fibrin, retraction of the blood clot), the whole process occurs synergistically. Many of the factors involved are enzymatic cofactors, and most of the reaction occurs on cell and platelet membranes (Figure 31-3). Many refinements in the understanding of blood coagulation have come about through study of “experiments of nature,” in which discrete defects of the clotting process have been identified in patients with bleeding diatheses, as illustrated by the factors and deficiency states listed in Table 31-1.
| INTERNATIONAL NUMBER OR TERM* | PLASMA FACTOR AND ALTERNATIVE NAMES† | CAUSE OR DESCRIPTION OF DEFICIENCY |
|---|---|---|
| I | Fibrinogen | Liver disease |
| II | Prothrombin | Liver disease or vitamin K deficiency |
| III | TF, thromboplastin | Deficiency of TF probably does not occur |
| IV | Ca++ | Never deficient without tetany |
| V | Proaccelerin | Parahemophilia, rare |
| VII | Proconvertin | Liver disease or vitamin K deficiency |
| VIII | Antihemophilic globulin, AHF A | Hemophilia A, 80% of hemophilics |
| IX | Christmas factor, AHF B | Hemophilia B (Christmas disease), depressed with vitamin K deficiency |
| X | Stuart-Prower factor | Liver disease or vitamin K deficiency |
| XI | Plasma thromboplastin antecedent, AHF C | Factor XI hemophilia (hemophilia C) |
| XII | Hageman factor | Generally no clinical symptoms but may have thromboses, rare |
| XIII | Fibrin-stabilizing factor, Laki-Lorand factor, fibrinase | Delayed bleeding, defective healing, rare |
| PF3 | Platelet factor 3 | Thrombocytopenia |
| — | Protein C | Liver disease or vitamin K deficiency |
| — | Protein S | Liver disease or vitamin K deficiency |
| — | Protein M | Liver disease or vitamin K deficiency |
| vWF | von Willebrand factor | vWD types I, IIa, IIb, IIc, III |
| Pre-K | Prekallikrein, Fletcher factor | |
| HMWK | High-molecular-weight kininogen |
AHF, Antihemophilic factor; TF, tissue factor.
* Roman numerals were assigned in 1958 by the International Committee on Blood Clotting Factors. Factor VI, originally assigned to prothrombin converting principle (prothrombinase), has since been abandoned.
† Most sources currently use factor numbers.
Initiation of coagulation after injury is a complex process involving an initial pathway of thrombin generation, which autocatalyzes a subsequent burst of additional thrombin generation sufficient to convert fibrinogen to fibrin (see Figure 31-3). Before the process is described, a brief review of the crucial factors and cofactors and how they function is warranted.
Vitamin K–dependent clotting factors
The amino terminus Gla domain is crucial for the lipid binding of these proteases to their substrate membranes. In the presence of seven Ca++ ions intercalated within the three-dimensional structure of the Gla domain, the protein undergoes a conformational change that places its hydrophilic domain at one end of the three-dimensional protein structure, with the hydrophobic moieties facing outward. This arrangement is crucial because it allows the protein to settle into the lipid membrane and exert its effects locally rather than systemically in the vasculature. Before these events can occur, however, the Gla residues must be formed post-translationally by carboxylation of their precursor glutamate residues by a specific γ-glutamyl carboxylase that requires the uncleaved preprotein leader sequence of amino acids to bind to the protein. This carboxylase enzyme requires oxygen, carbon dioxide, and vitamin K to function (see Figure 31-8). For every glutamate residue carboxylated, one molecule of reduced vitamin K is converted to its epoxide form. A separate enzyme, vitamin K epoxide reductase, converts the vitamin K back to the reduced form. This reductase is the target of the warfarin-like anticoagulants and is discussed in greater detail later.
Regulation of Coagulation
When discussing hemostatic mechanisms, consideration should be given to the natural inhibitors of blood clotting. As important as the procoagulant process is, it is equally important to ensure that inappropriate clotting does not occur. The intent of the clotting system is to seal a site of vascular compromise; powerful antithrombotic mechanisms must come into play to ensure that clotting remains limited to the injured area. Several mechanisms of antithrombosis have been elucidated; they are discussed in detail subsequently and summarized in Figure 31-4. At the heart of the matter is how to control the extremely efficient clotting cascade after it is initiated.
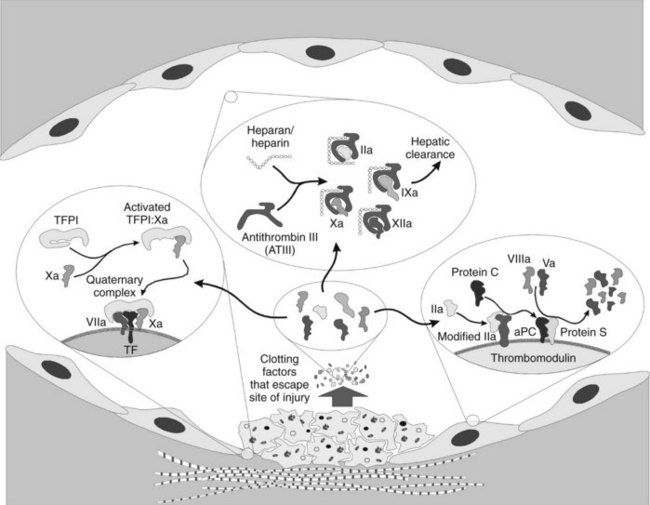
Thrombomodulin, as the name implies, alters the conformation of the thrombin and effectively removes its ability to cleave fibrinogen, activate platelets, and activate factors V and VIII. Instead, the new conformation of thrombin imparts a 2000 times greater affinity for activation of the vitamin K–dependent protein C.12 Activated protein C (aPC) has considerable homologous characteristics with the other vitamin K–dependent factors, complete with a Gla domain, hydrophobic domain, and active serine protease domain. The cofactor for aPC is protein S, a membrane-bound protein that has no inherent activity. When aPC is bound, the complex efficiently cleaves and destroys any factors Va and VIIIa that might have been liberated from the platelet surfaces, however, slowing coagulation and protecting the normal individual against random intravascular coagulation.
PROCOAGULANT AGENTS
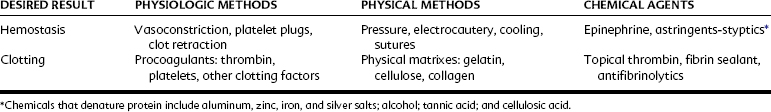
In medical and dental practice it is essential to take appropriate precautions to avoid serious hemorrhage. This admonition is particularly true for patients with hemophilia, patients with hematopoietic disease, and patients receiving therapies known to affect hemostasis. Precautions, which may include the administration of clotting factors or hospitalization or both, are prudent in these cases. In contrast, normal patients usually require no more than temporary hemostatic assistance (e.g., pressure packs, hemostatic forceps, ligation, or other locally active measures) to facilitate normal hemostasis and allow clotting to occur. Table 31-2 outlines various methods for controlling bleeding.
Topically Applied Clotting Factors
Fibrin sealant, also sometimes referred to as fibrin glue, is one of the more promising hemostatic aids to appear in recent years.5 With this agent, the concept of the application of topical thrombin is taken one step further. Bovine or human thrombin and calcium chloride are mixed in one of two syringes; purified human fibrinogen with factor XIII, aprotinin, and other plasma proteins (fibronectin and plasminogen) are in the second syringe. The two solutions are mixed in a single delivery barrel, where the thrombin cleaves the fibrinogen to fibrin monomers. Initially, they are gelled by hydrogen bond formation, but in 3 to 5 minutes the factor XIII in the presence of Ca++ initiates cross-linking and increases the tensile strength of the clot.5 As the clot solidifies, the sealant becomes milky white.
Fibrin sealant is commercially available in the United States. The protein fractions are lyophilized and require careful reconstitution at 37° C under sterile conditions; proper mixing of the materials requires approximately 30 minutes to perform. As a result, the emergent use of this material is difficult; typically, it is used more in planned surgeries in patients with known bleeding disorders. It is also an expensive medication; 1 mL of the material costs several hundred dollars. Fibrin sealant works well, however, in stopping the microbleeding and oozing that often accompany dental procedures.27
Astringents and Styptics
The terms astringents and styptics are interchangeable, referring to different concentrations of the same drugs. Many chemicals have vasoconstrictive or protein-denaturing ability, but relatively few are appropriate for dentistry. The suitable preparations are primarily salts of several metals, particularly zinc, silver, iron, and aluminum. Aluminum and iron salts are quite acidic (pH 1.3 to 3.1) and irritating.36 Iron causes annoying, although temporary, surface staining of the enamel, whereas silver stains may be permanent.
Currently, astringents are generally used in dentistry only to aid hemostasis while retracting gingival tissue. Other applications, such as controlling bleeding after surgery, are not looked on as favorably as in the past, when 20% ferric subsulfate (Monsel’s solution) and 8% zinc chloride were among the most popular agents used. Aluminum and iron salts function by denaturing blood and tissue proteins, which agglutinate and form plugs that occlude the capillary orifices. In a rabbit mandible model, when ferric sulfate salts were left in an osseous wound, there was an intense foreign body reaction and delayed healing in many of the experimental sites compared with the control sites.24 When the salts were irrigated, and the coagulum was curetted away, this response was markedly diminished with no persistent inflammation or delay of osseous repair.
Systemic Measures
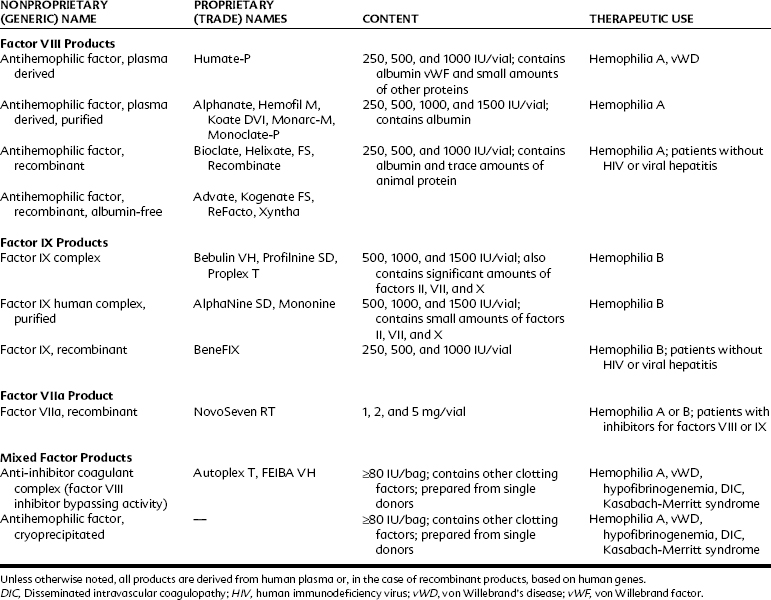
Patients with acquired or genetic bleeding disorders usually have deficiencies in platelet number, platelet function, or faulty or missing clotting factors. Bleeding may develop several hours after trauma or surgery. Uncontrolled bleeding does not generally appear with superficial abrasions, but hemarthrosis and hemorrhage are common with deeper injuries. Thrombocytopenia is frequently drug-induced or associated with other myelogenous diseases; hemophilia disorders are generally inherited. With proper evaluation and supportive therapy (Table 31-3), extensive surgery can usually be accomplished without serious incident.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


