CHAPTER 30 Antianemic and Hematopoietic Stimulating Drugs
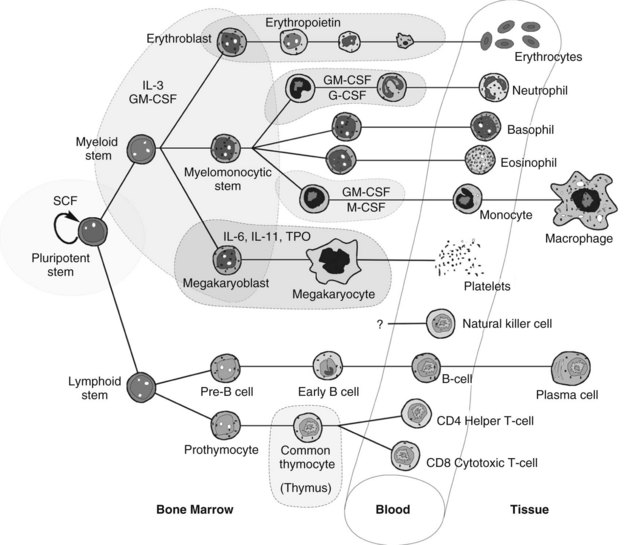
Hematopoiesis is the intricate system of growth and differentiation of immature pluripotent/multipotent stem cells into all the formed elements of the blood (Figure 30-1). These stem cells, derived embryologically in the liver and later from bone marrow,33 divide early in development into either myeloid or lymphoid precursors. The myeloid precursors differentiate into the erythrocytes, megakaryocytes (which give rise to thrombocytes [platelets]), neutrophils, and monocytes. The lymphoid precursors give rise to the T-cell and B-cell lymphocytes, natural killer cells, and all their respective subtypes. The derivation of eosinophils and basophils lies in the myeloid stem line, but appears to be downstream of the common myeloid precursors.26 Hematopoietically active bone marrow retains essentially the same mass throughout life, and although cell-producing bone marrow is found in practically all bones through adolescence, it becomes restricted to the vertebrae, sternum, ribs, pelvis, scapulae, parts of the skull, and epiphyseal ends of the long bones after approximately age 20 years.
ANEMIA
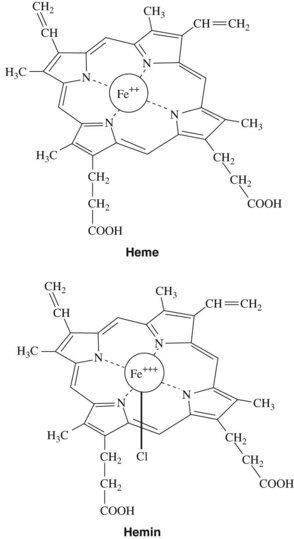
After anemia has been detected, it can be characterized by evaluating the hemoglobin in the erythrocytes. Normal amounts of hemoglobin per unit volume of blood (assayed on peripheral blood draw) are 15.2 ± 2.2 g/dL for men and 13.7 ± 2.1 g/dL for women. Hemoglobin is the oxygen-carrying component of red blood cells. It comprises three components: iron, porphyrin rings, and globin chains. Alterations in any one of these three components can be a cause for a clinical anemia. In normal hemoglobin, iron in the ferrous form (Fe++) is chelated into the middle of the porphyrin chemical ring to yield heme, the nonprotein component of hemoglobin (Figure 30-2).
Hemoglobin accounts for approximately 95% of the dry weight of mature erythrocytes. Any significant changes in hemoglobin are often directly reflected in the way the erythrocytes look or behave grossly. Classically, laboratory analyses for anemia have reviewed erythrocyte size, shape, and color intensity. The size is determined by the mean corpuscular volume (MCV). The normocytic, or normal size, range is 80 to 100 fL/cell. Cells that are too small are termed microcytic, whereas cells that are too large are termed macrocytic. The shape of the red blood cells is also important in diagnosing the cause of anemia. Box 30-1 lists terms that describe various shapes found on a peripheral blood smear. The color intensity of the cell is reflected in the mean corpuscular hemoglobin (normally 26 to 34 pg/cell) and the mean corpuscular hemoglobin concentration (normally 31 to 36 g/dL). These two parameters, along with MCV, collectively referred to as the red blood cell indices, are extremely helpful in delineating the causes of a particular anemia.
BOX 30-1 Descriptive Terms of Red Blood Cell Morphologic Characteristics
| TERM | DESCRIPTION |
|---|---|
| Poikilocytosis | Irregular erythrocyte shape |
| Anisocytosis | Irregular erythrocyte size |
| Polychromasia | Change in amount of hemoglobin |
| Sickling | Sickle cell disease and trait |
| Targeting | “Bull’s-eye” look to the erythrocytes caused by hemoglobin C and liver disease |
| Leptocytes | Hemoglobin in the border with pigmentation in the center; found in thalassemia, obstructive jaundice, any hypochromic anemia, hemoglobinopathy, and after splenectomy |
| Spherocytes | Round erythrocytes (not biconcave), caused by hereditary spherocytosis or by immune or microangiopathic hemolysis |
| Schistocytes | Fragments of erythrocytes; found in hemolytic transfusion reactions, microangiopathic hemolysis, and other severe anemias |
| Acanthocytes | Distorted (“thorny”) erythrocytes with protoplasmic projections; seen in severe liver disease and with high titers of bile, fats, or toxins |
| Howell-Jolly bodies | Smooth, round remnants of nuclear chromatin; seen in megaloblastic and hemolytic anemias and after splenectomy |
| Nucleated erythrocytes | Found in severe bone marrow stress (e.g., hemorrhage, hemolysis), marrow replacement by tumor, extramedullary hematopoiesis |
The various kinds of anemia are classified by their typical effect on the erythrocytes (Table 30-1). When anemia results from a loss of blood (intrinsically from hemolysis or extrinsically from hemorrhage) or because of a decrease in production of normal erythrocytes, the cells are still normal, just fewer in quantity. These anemias are normocytic and normochromic. When anemia is caused by a decrease in the production of properly formed hemoglobin, the cells tend to be smaller (because hemoglobin comprises such a high percentage of erythrocyte content) and paler in color. These forms of anemia are known as microcytic and hypochromic and are usually the result of defective or inadequate iron absorption. Forms of anemia that cause the red blood cells to mature incompletely and retain some DNA content result in larger cells; these are known as macrocytic or megaloblastic anemia. They generally occur as a result of a deficiency in vitamin B12, folic acid, or both nutrients. In these forms of anemia, the cells may also have a darker or hyperchromic color.

Iron and Iron Deficiency Anemia
Nutrition and physiologic characteristics
Iron deficiency anemia is the most common cause of anemia worldwide and may occur for many reasons: inadequate nutrition in relation to rate of growth (qualitative or quantitative); defective absorption, transport, or storage (e.g., congenital atransferrinemia or inability to release iron from transferrin to the red blood cells and their precursors); or blood loss from hemorrhage (most commonly gastrointestinal), menstruation, or blood donation. In the United States, iron deficiency anemia is found in 7% of infants, 4% to 5% of children, and 9% to 16% of menstruating women.5 Only 2% to 3% of men have iron deficiency anemia, and women taking oral contraceptives tend to have lower rates because progestins reduce menstrual blood loss. Children 6 months to 2 years old are particularly vulnerable because of their high growth rate coupled with weaning off breast milk and onto cow’s milk. Cow’s milk is low in absorbable iron and may irritate the intestines. Pregnancy may precipitate iron deficiency anemia by rapidly increasing the blood volume, sometimes requiring two to five times the normal intake of iron. According to a World Health Organization technical report,17 women who have sufficient iron reserves to support the increase in hemoglobin production during pregnancy and who breastfeed their infants are generally capable of meeting their iron needs by diet alone, although supplementation is still recommended. In a nonpregnant, normal, healthy individual, iron reserves and recycling are so effective that even extreme reduction of iron intake may be insufficient to cause severe anemia.12
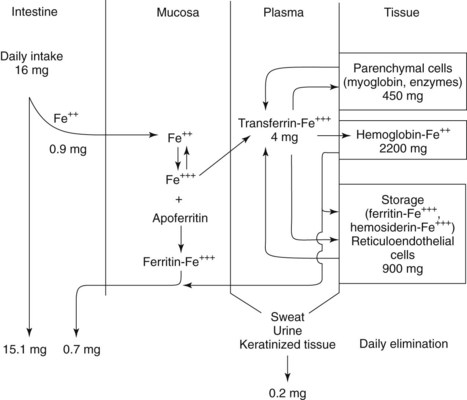
Men average 3.8 g total iron (50 mg/kg) and women average 2.3 g (35 to 42 mg/kg). Approximately 60% to 80% of the iron in the body is incorporated into hemoglobin (Figure 30-3). Anemia is the primary presenting sign of iron deficiency. Approximately 10% to 25% is sequestered in reticuloendothelial cells in the storage forms ferritin and hemosiderin (described later), and another 10% to 15% is associated in parenchymal cells with myoglobin. Less than 1% is used in various enzymes, most notably the cytochromes, and trace amounts are linked to the plasma transport protein transferrin. The amount of stored iron varies with intake and demand, averaging 400 mg in women and 1000 mg in men.
Iron therapy
The intuitive treatment of any disease state that is accompanied by extreme fatigue, weakness, and loss of color includes increased dietary intake, and the ancient Greeks, Hindus, and other early peoples turned to iron in many forms simply because it represented “strength.” Although Sydenham is generally credited with the first rational use of iron (iron filings in wine) for treating anemia in 1681, it was not known that iron was actually present in blood until 30 years later, when Lemery and Geoffry demonstrated its presence. Shortly thereafter, Menghini, an Italian physician, showed that foods with iron actually increase blood iron, but it was not until approximately 1830 that a pill containing iron (ferrous sulfate and potassium carbonate, 1 : 1) was introduced into medicine by Blaud,3 an event that marked the beginning of modern treatment of iron deficiency anemia.
PORPHYRIA
Porphyrin is produced in an eight-step process that occurs in the mitochondria and in the cytosol. The two principal cell types involved are the developing erythroblasts and reticulocytes of the bone marrow (mature erythrocytes lack mitochondria and are unable to synthesize porphyrin) and the liver hepatocytes. As a result, two general classifications of porphyria exist—erythropoietic and hepatic—that are divided further into nine varieties (Table 30-2), each corresponding to a particular enzyme deficiency in the synthetic pathway of porphyrin. These deficiencies may be genetic in nature or caused by medications.
| PORPHYRIA | SITE OF EXPRESSION | PRINCIPAL CLINICAL FEATURE |
|---|---|---|
| Acute intermittent porphyria | Liver | Neurologic |
| δ-Aminolevulinic acid dehydratase deficiency porphyria (rare) | Liver | Neurologic |
| Hereditary coproporphyria | Liver | Neurologic, photosensitivity |
| Porphyria cutanea tarda | Liver | Photosensitivity |
| Variegate porphyria | Liver | Neurologic, photosensitivity |
| Hepatoerythropoietic porphyria | Liver, bone marrow | Photosensitivity |
| Congenital erythropoietic porphyria | Bone marrow | Photosensitivity |
| Erythropoietic protoporphyria | Bone marrow | Photosensitivity |
| X-linked sideroblastic anemia | Bone marrow | Hemolytic anemia |
Several events can precipitate an attack. Physiologic stressors, such as surgery, excessive alcohol intake, illnesses, and infections, may induce hepatic heme oxygenase, which breaks down heme. Endocrine changes, such as may occur around a woman’s menses, or synthetic estrogens and progestins may also induce an attack. More than 1000 medications have been categorized with regard to their porphyrinogenicity,32 of which a few reactions are well documented and many are still anecdotal. What is accepted is that endocrine properties of the drug, affinity for cytochrome P450, hepatic load, and capacity to modulate nuclear receptors affecting gene transcription (particularly 5-aminolevulinate synthase [ALAS1]) all play a role in how porphyrinogenic a drug might be.
Several medications commonly used in dentistry and medicine (Box 30-2) are steroid-based or metabolized by, and induce the synthesis of, the cytochrome P450 enzyme system, which leads to increased accumulation of porphyrin precursors.22 The response of any individual to any of these medications can be highly variable; the proposed unsafe medications should be discussed with the physician on a case-by-case basis. In susceptible porphyric patients, which also includes individuals with hereditary coproporphyria and variegate porphyria, dose reductions or avoidance of specific medications may be necessary. Thunell and colleagues32 have proposed a standardized method to determine a drug’s probable porphyrinogenicity, and an Internet database is available at http://www.drugs-porphyria.org. Poor nutritional intake has also been associated with acute attacks.
| SAFE | POSSIBLY UNSAFE |
|---|---|
| Acetaminophen | Alcohol |
| Amitriptyline | Alkylating agents |
| Aspirin | Barbiturates (severe) |
| Atropine | Carbamazepine |
| Chloral hydrate | Chlordiazepoxide |
| Clorazepate | Chlorpropamide |
| Diazepam | Chloroquine |
| Digoxin | Clonidine |
| Diphenhydramine | Dapsone |
| Glucocorticoids | Ergots |
| Guanethidine | Erythromycin |
| Hyoscine | Estrogens, synthetic |
| Ibuprofen | Food additives |
| Imipramine | Glutethimide |
| Insulin | Griseofulvin |
| Labetalol | Hydralazine |
| Lithium | Ketamine |
| Naproxen | Meprobamate |
| Nitrofurantoin | Methyldopa |
| Opioid analgesics | Metoclopramide |
| Penicillamine | Nortriptyline |
| Penicillin and derivatives | Pentazocine |
| Phenothiazines | Phenytoin |
| Procaine | Progestins |
| Propranolol | Pyrazinamide |
| Selective serotonin reuptake inhibitors |
Management of acute intermittent porphyria has been primarily aimed at avoiding exacerbating conditions. Adequate caloric intake, prompt diagnosis and treatment of infections (including odontogenic and other orofacial infections), and care in not taking medications known to trigger attacks are strategies the patient can use to minimize the risk of developing a crisis. In patients who have photoreactive porphyria, avoidance of sunlight, wearing clothing to cover the skin, and generous use of sunscreen lotion are helpful. If an acute attack occurs that is not amenable to glucose infusion, a medication of choice is lyophilized hemin with sodium carbonate. Hemin is ferric heme that has a Cl− on one of the two available coordination sites for Fe+++ (see Figure 30-2). On mixing with sterile water, hemin is converted in the resulting alkaline solution to hematin by replacement of the Cl− with an OH− group. Hematin serves as an enzymatic inhibitor of porphyrin synthesis by decreasing the concentration of the precursors porphobilinogen and δ-aminolevulinic acid. The reconstituted drug is unstable, however, and has been frequently associated with thrombophlebitis and increased coagulopathy. Palliative use of opioid analgesics is also often indicated during porphyric exac/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


