CHAPTER 29 Lipid-Lowering Drugs
Because the deposition of cholesterol in arteries is a defining feature of atherosclerosis, strategies for its prevention and treatment include methods to reduce plasma cholesterol. Dentists need to understand lipid-lowering drugs as the overall dental patient population of the United States matures, and increasing numbers of patients take these drugs for prevention and therapy of atherosclerosis. Dentists also need to follow the development of the cholesterol synthesis inhibitors because of their implication in the stimulation of bone formation through inhibited isoprenoid synthesis leading to decreased osteoclast activity and increased osteoblast differentiation.23,28
Guidelines on patient cholesterol management are issued periodically by the Adult Treatment Panel of the National Cholesterol Education Program (NCEP).18 More recent guidelines have included recommendations for improved lifestyle changes, such as increasing exercise and decreasing consumption of saturated fat and cholesterol, which improve cholesterol levels in some patients without drug therapy.
CHOLESTEROL AND ATHEROSCLEROSIS
Atherosclerosis is caused by development of fatty streaks and plaques in large and medium-sized arteries, especially the aorta, coronary arteries, carotid arteries, renal arteries, and arteries of the legs. Plaques develop in the intimal wall of the vessels after deposition of cholesteryl esters derived from certain lipoproteins. The particular clinical manifestation of atherosclerosis depends on the degree to which the lesions have progressed in a particular part of the vasculature. The presence of atheromatous lesions can have several effects on the circulation to a prescribed area: (1) blood flow may be obstructed by the plaques themselves or by associated thrombi, (2) vascular reactivity and control of blood flow may be lost, and (3) vessels may become weakened and subject to rupture. Observations of initial lesions in young children have shown that this disease begins at an early age and progresses gradually, with clinical symptoms generally appearing much later in life.35,43
Lipoprotein Metabolism
Classification of plasma lipoproteins is based on the density of the complexes, with a lower density indicating higher lipid content (Table 29-1). Chylomicrons are the largest particles and possess the greatest proportion of lipid. The other lipoproteins are very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), low-density lipoprotein (LDL), high-density lipoprotein (HDL), and lipoprotein(a). Two separate lipoprotein-producing systems or metabolic pathways are responsible for the transport of lipids (Figure 29-1): the exogenous pathway, producing chylomicrons in the intestinal mucosa primarily from dietary fat, and the endogenous pathway, producing VLDL in the liver with triglycerides from hepatic metabolism of dietary carbohydrates.
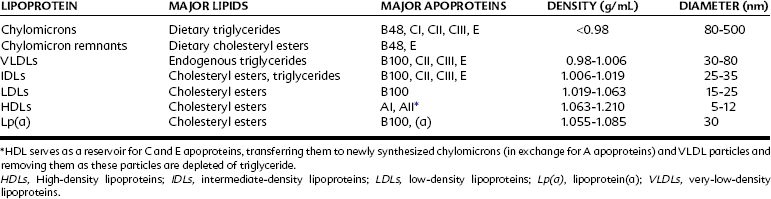
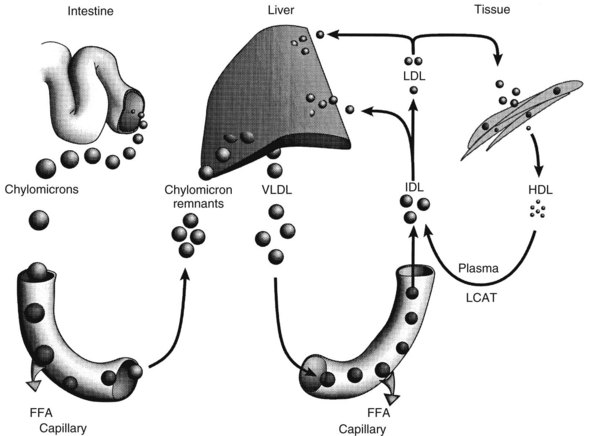
Apoprotein CII serves as a stimulus for endothelial lipase activity. The remaining chylomicron remnants, depleted of triglycerides and CII but relatively enriched in cholesterol and apoproteins B48 and E, are released back into the circulation. The exogenous pathway terminates in the liver, where the chylomicron remnants are actively taken up by hepatocytes. Apoproteins B48 and E help trigger this receptor-mediated endocytosis. Within the liver, the remnants are digested, releasing the remaining lipids for further metabolism (Figure 29-2).
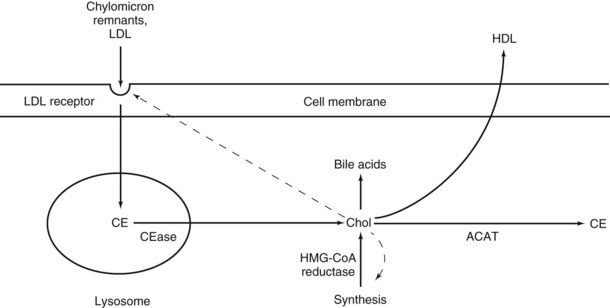
Enterohepatic cycling includes the following processes36: (1) synthesis of cholesterol and conversion to bile acids by the liver, (2) secretion of biliary cholesterol and bile acids into the small intestine, (3) absorption of bile acids from the terminal ileum, and (4) transfer through the portal system and reuptake by the liver. Reverse cholesterol transport is the process by which HDL synthesized by the liver and intestine takes up cholesterol from peripheral cells and transports it back to the liver. HDL cholesterol is sometimes referred to as the “good” cholesterol because it represents the amount of cholesterol being removed from most of the body and because higher levels of HDL cholesterol are associated with decreased risk of atherosclerosis. HDL also assists in the removal of triglycerides from the bloodstream by delivering to chylomicrons and VLDL the C and E apoproteins necessary for their processing.
Role of Lipoproteins in Atherosclerosis
The presence of cholesterol as an integral component of arterial plaques has been known for decades, and many studies have been done to determine whether abnormal concentrations of cholesterol are involved in the origin or progress of plaque formation. These studies have shown that plasma cholesterol concentrations are higher in patients with coronary artery disease than in normal patients, and that the relationship between serum cholesterol concentration and risk of premature death from coronary artery disease is continuous and graded.38,42 More recent studies of cholesterol reduction therapy in patients with hyperlipidemia have shown that reduction of plasma cholesterol can cause regression of plaque formation and decrease the mortality rate from atherosclerosis.18,44
An increase in plasma LDL may be caused either by overproduction of its VLDL precursor or, more commonly, by retarded clearance of LDL from the blood. The plasma half-life of LDL is much longer than the half-lives of VLDL and IDL. Although LDL receptor–dependent uptake predominates normally, only the alternative phagocytic pathway is available in patients with severe familial hypercholesterolemia, who congenitally lack functional LDL receptors.7 LDL contains most cholesterol in the plasma, and it is the lipoprotein group most directly associated with coronary heart disease.27,41 High plasma concentrations of HDL are inversely related to the risk of coronary heart disease.10,41
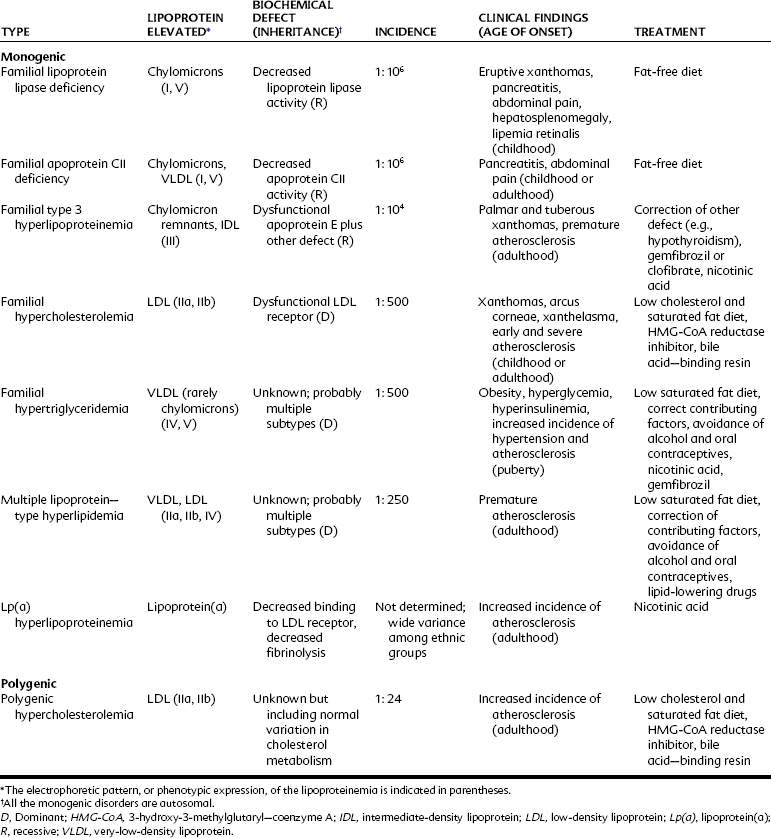
Hyperlipidemia may be primary (i.e., genetic in origin) or result from dietary factors; disease states such as diabetes mellitus, hypothyroidism, or uremia; drugs such as alcohol, oral contraceptives, or glucocorticoids; or a combination of causes. A summary of various types of primary hyperlipidemias is provided in Table 29-2.
Risk Factors in Atherosclerosis
In addition to high plasma cholesterol, several risk factors have been identified with an increased incidence of atherosclerosis, including hypertension, cigarette smoking, sedentary habits, obesity, diabetes mellitus, hypothyroidism, male gender, and a family history of atherosclerosis or diabetes. As shown in Table 29-2, the clinical manifestations of genetic or familial hyperlipoproteinemias are severe. The lipid-lowering agents discussed in this chapter have proved quite useful in treating many of these disorders and even in reversing the progression of atherosclerosis in patients without specific metabolic defects in lipid metabolism.
Treatment Guidelines From the National Cholesterol Education Program
The primary guidelines from NCEP for treatment of patients with hyperlipidemia are to increase exercise, decrease saturated fat and cholesterol in the diet, and lower elevated LDL-cholesterol levels using drugs.18 Diabetes also predicts a high risk for coronary heart disease, and these patients benefit from treatment with hypolipidemic drugs. Diabetic patients usually have elevated triglycerides and low HDL cholesterol, requiring different types of drug treatment. Diabetic patients who have confirmed coronary heart disease are at even greater risk and need more specific, aggressive treatment to decrease risk of mortality.
THERAPEUTIC AGENTS
Therapy with drugs that reduce plasma cholesterol is used to delay or reverse the progression of atherosclerosis and decrease the mortality and morbidity rates from the associated clinical manifestations of this disease. Strong evidence supports the idea that correction or lowering of plasma lipid concentrations is beneficial in many instances.18,41 These drugs are helpful in treating many familial hyperlipidemias, and they are recommended for use in patients with hyperlipidemias of secondary etiology that cannot be corrected by other means. Lipid-lowering drugs are most frequently administered to patients with a history of ischemic heart disease in an attempt to avoid future fatal episodes of myocardial infarction.
The goal of therapy is to reduce lipid levels as much as possible without producing metabolic derangements or adverse drug effects.38,42 Altering the diet is generally the initial therapeutic measure, along with correcting any disease state or condition contributing to hyperlipidemia. If nonpharmacologic therapy is insufficient, drug administration should be considered depending on determinants such as age, gender, presence of ischemic vascular disease, and coexistence of other risk factors. Even when pharmacologic antihyperlipidemic treatment is instituted, however, nonpharmacologic management, including reduction of dietary cholesterol and saturated fatty acids, weight reduction, exercise, and smoking cessation, remains the cornerstone of therapy.
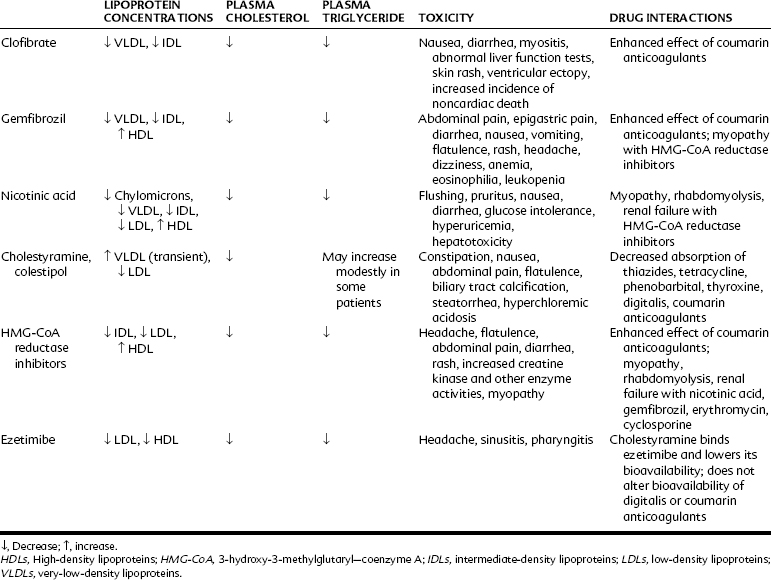
The following hypolipidemic agents are considered in this chapter: (1) derivatives of fibric acid, including clofibrate and gemfibrozil; (2) nicotinic acid; (3) the bile acid sequestrants cholestyramine and colestipol; (4) inhibitors of HMG-CoA reductase; (5) cholesterol absorption inhibitors; and (6) other agents. The effects of the major drugs on the various classes of lipoprotein are listed in Table 29-3.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


