Part 4: Appliance Techniques
28
Biological responses during orthodontic tooth movement
UPDATE ON BONE PHYSIOLOGY
Bone remodelling associated with orthodontic tooth movement has become clearer with an increased knowledge of osteoblast and osteoclast interactions. Bone comprises a matrix of mineral (70%) based on a type 1 collagen scaffold (27%). The remaining 3% of the matrix comprises minor collagens and other proteins including osteocalcin, osteonectin, osteopontin, phosphoproteins, sialoproteins, glycoproteins, lipids and glycosaminoglycans. The matrix also contains a wide variety of polypeptide growth factors. Many of these are bound to proteins (binding proteins) that render them inactive. These growth factors are liberated from the matrix as well as the binding proteins by osteoclast acid hydroxylases. Within the matrix are blood vessels and these features are important in cellular actions and bone function. The main bone cell types are osteoblasts, osteocytes and osteoclasts.
Osteoblasts
Osteoblasts arise from undifferentiated mesenchymal cells which differentiate through an immature pre-osteoblast state to the mature and functional osteoblast state. In this mature state, the osteoblast synthesises both collagenous and non-collagenous bone proteins which constitute the organic matrix or osteoid. Within this osteoid, type I collagen accounts for 90% of the protein in bone. In addition to this bone-forming activity, the osteoblast is responsible for osteoclast recruitment and activation in response to hormonal and mechanical challenges. It is the osteoblast that has receptors for most of the bone-resorbing hormones such as parathyroid hormone (PTH) and the cytokines. This is important when osteoblast and osteoclast interactions are considered further (see below).
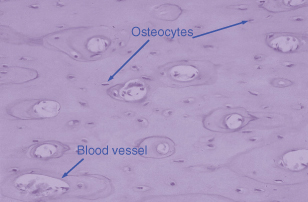
Osteocytes represent terminally differentiated osteoblasts, and are cells that have produced sufficient matrix to encase themselves within bone. They remain connected to other osteocytes and the surface-lining osteoblasts through canaliculi. Entombed within a fairly rigid matrix, they are ideally positioned to detect any mechanical forces that might be placed on bone such as in orthodontic tooth movement. The osteocytes can then send signals to the osteoblasts, which may recruit and activate osteoclasts to cause bone resorption. Figure 28.1 shows the position of osteocytes within the bone, each lying within its own lacunae.
Figure 28.1 Haematoxylin and eosin section of bone illustrating the position of osteocytes within a calcified matrix. The large lacunae are blood vessels and both the osteocyte and the blood vessel are clearly marked.
Osteoclasts
Osteoclasts are the main bone resorption cells. Their appearance in culture is striking, and each has a large number of nuclei and a highly active peripheral cytoplasm. The latter is a crucial morphological feature since it allows the osteoclasts to ‘seal’ off a portion of bone matrix and then create an acid environment for the dissolution of bone as well as the cleavage of growth factors from their binding proteins. These growth factors are usually cleaved in a latent (inactive) form but the acidity will release the growth factors from this latency, enabling them as active growth factors.
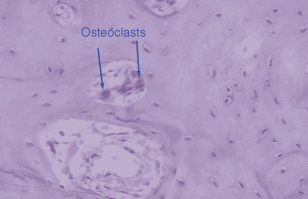
Osteoclasts are formed by fusion of circulating monocytes in response to specific signals from the osteoblasts. Figure 28.2 shows very clearly the importance of this vascular origin. Within the blood vessel are several osteoclasts and these can be recruited to specific regions of resorptive activity. These osteoblast–osteoclast interactions have been an area of exciting discovery in the past few years. The osteoclast by contrast to the osteoblast has very few receptors for hormones, e.g. calcitonin and retinoic acid. Most of the hormones that the osteoclast has receptors for have inhibitory actions (e.g. prostaglandin E2) and these result in a decrease of osteoclast activity. A number of hormones elevate intracellular cyclic AMP levels and this second messenger seems to be a major pathway for inhibition of osteoclast activity.
Figure 28.2 The lineage of osteoclasts is important. This haematoxylin and eosin section demonstrates a blood vessel in bone and within the blood vessel three osteoclasts, two of which are clearly marked.
Osteoblast–osteoclast Interactions
Osteoclasts in isolated culture have relatively little activity and are not stimulated with bone-seeking hormones such as PTH or cytokines. It is only when co-cultured with osteoblasts and bone-seeking hormones that osteoclasts start to demonstrate motility or to resorb calcified matrices such as bone or dentine. Thus osteoclasts cannot resorb bone without prior activation by osteoblasts but for many years what these signals comprised was not known. Furthermore the control mechanisms and feedback loops to control the bone remodelling could only be guessed at. Recently a number of important discoveries have helped clarify the situation.
The first significant step was the recognition that three members of the tumour necrosis factor (TNF) and TNF receptor superfamily are involved in the formation and activation of osteoclasts. A well-known step in the formation of osteoclasts is the action of the macrophage colony-stimulating factor (M-CSF) which stimulates differentiation and proliferation of haemopoietic progenitors or pre-osteoclasts. These pre-osteoclasts express a receptor called RANK (receptor activator of nuclear factor [NF]-κB) and the ligand for this receptor (RANKL) is produced by osteoblasts. RANKL is important for the differentiation of osteoclasts from monocytic precursors as well as maintaining osteoclast function.
Unfortunately, a feedback loop makes this a bit more complicated. Osteoblasts also produce and secrete osteoprotegerin (OPG), which acts as a decoy receptor that can block RANKL/RANK interactions. Bone-resorbing hormones can increase RANKL expression in osteoblasts, and some can also decrease OPG expression.1 Bone cells appear to express the membrane-bound form of RANKL, and, thus, osteoblasts must physically interact with osteoclast precursors in order to activate RANK. This complex picture can be seen clearly in studies where, in mice, the genes for OPG are manipulated so that OPG is overexpressed. Since OPG blocks RANKL/RANK interactions, osteoclast function is diminished and excess bone is laid down. The mouse becomes osteopetrotic. This osteopetrotic state can be reversed by injection of the RANKL protein. Similarly the gene for OPG can be deleted in mice with a resultant increase in osteoclast activity and the development of an osteoporotic state, sufficiently severe to cause bone fractures.2 Potentially all of these findings could provide information to develop a strategy to deal with the consequences of osteoporosis, osteopetrosis and other crippling diseases. No doubt other pathways which modulate osteoblast and osteoclast interactions will be found and modify our thinking on bone remodelling. These complex interactions are summarised in Figure 28.3 and the osteoblast, osteoclast and osteocyte relationships in Figure 28.4.
Figure 28.3 The ‘missing link’ between osteoblasts and osteoclast interactions can be explained in part through RANK ligand (RANKL). Osteoblasts produce RANKL in response to stimuli such as mechanical stress or hormones such as parathyroid hormone (PTH). RANKL is responsible for formation of osteoclasts from haemopoietic progenitor cells and also for increasing the activity of osteoclasts and thus increasing bone resorption. M-CSF, macrophage colony-stimulating factor.
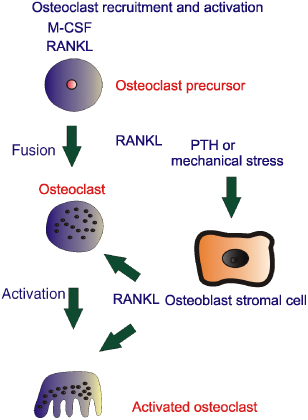
Figure 28.4 Osteoblasts have most of the receptors for bone-seeking hormones, some of which are shown within the schematic. In response to these hormones or indeed mechanical stress, RANK ligand (RANKL) is produced, which is responsible for recruitment and activation of osteoclasts. The osteoblast in response to />
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


