CHAPTER 24 Management of the Medically Compromised Patient: Hematologic Disorders, Cancer, Hepatitis, and AIDS
HEMOPHILIA
DISORDERS OF HEMOSTASIS
The hemophilias are disorders of hemostasis resulting from a deficiency of a procoagulant. Hemophilia is an inherited bleeding disorder affecting approximately 1 in 7500 males.1 Hemophilia A, or classic hemophilia, is a deficiency of factor VIII, also known as antihemophilic factor. Factor VIII deficiency is the most common of the hemophilias and is inherited as an X-linked recessive trait. Therefore males are affected, females are carriers, and there is no male-to-male transmission. If a normal male has children with a carrier of hemophilia, there is a 50% chance that hemophilia will occur in each male offspring and a 50% chance that each female offspring will be a carrier. If a male hemophilic has children with a normal female, all male offspring will be normal, and all female offspring will be carriers. Hemophilia B, or Christmas disease, is caused by a deficiency of factor IX (plasma thromboplastin component) and is also inherited as an X-linked recessive trait. Factor IX deficiency is one-fourth as prevalent as factor VIII deficiency.2,3
Factor XI (plasma thromboplastin antecedent) deficiency, also referred to as hemophilia C or Rosenthal’s’ disease, is inherited as an autosomal recessive trait, with male and female offspring equally affected. This disorder is most frequently observed in those of Ashkenazi Jewish descent. Other factor deficiencies, such as those of factors II, V, and XIII (one case per 1 million population) and factor VII (one case per 500,000 population) are rare and are inherited as autosomal recessive traits.4,5
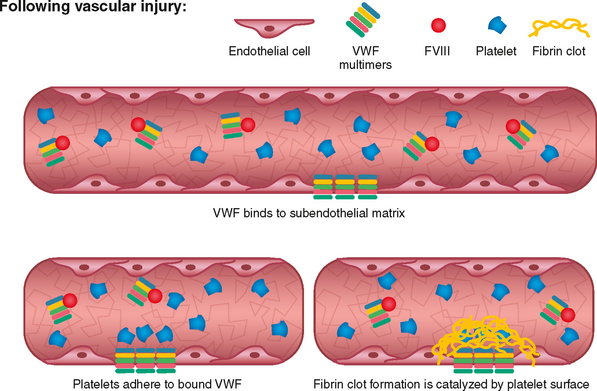
Von Willebrand disease is a hereditary bleeding disorder resulting from an abnormality of the Von Willebrand factor (VWF) found in plasma, platelets, megakaryocytes, and endothelial cells. VWF circulates in conjunction with factor VIII and is important in platelet adhesion to the subendothelium via collagen and therefore in the formation of the primary platelet plug. In von Willebrand disease, the VWF may have a quantitative or qualitative abnormality. The VWF is composed of subunits called multimers. Von Willebrand disease is divided into subtypes based on the platelet and plasma multimeric VWF structure. Optimal treatment of this disorder is dependent upon the subtype.6
Impaired formation of the platelet plug may result in bleeding from the skin and mucosa, bruising, epistaxis, prolonged bleeding after surgical procedures, and menorrhagia (Fig. 24-1). This is in contrast to hemophilia involving deficiencies of factors VIII and IX, in which the hallmark bleeding events involve muscles and joints (Fig. 24-2).
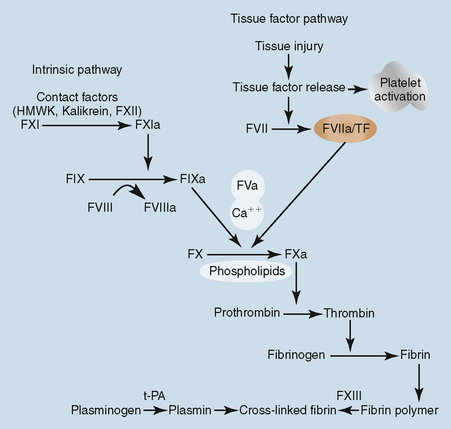
PROCOAGULANT CLASSIFICATION
Patients with severe deficiency may experience frequent bleeding episodes, often occurring two to four times per month. Bleeding episodes may be spontaneous, without a specific history of injury or trauma. Common sites of bleeding include joints, muscles, and skin. Hemarthroses (joint hemorrhages) are common, and symptoms include pain, stiffness, and limited motion. Repeated episodes of hemarthroses or muscle hemorrhage result in chronic musculoskeletal disease and culminate in debilitating painful arthritis. Commonly affected joints include knees, elbows, ankles, hips, and shoulders. Pseudotumors (hemorrhagic pseudocysts) may occur in several locations including the jaw, in which case curettage is indicated.7,8
Mouth lacerations are a common cause of bleeding in children with all severities of hemophilia. Sonis and Musselman evaluated 132 patients with factor VIII–deficient hemophilia and noted that “persistent oral bleeding resulted in the diagnosis of 13.6% of all cases of hemophilia.”9 About 29% of cases of mild hemophilia observed were discovered as a result of bleeding from the oral cavity. Of the cases diagnosed secondary to oral bleeding, 78% were the result of bleeding from the maxillary frenum, and 22% resulted from tongue bleeds. Thus initial diagnosis of hemophilia, especially in moderate or mild disease, may directly involve the dentist.
TREATMENT
The mainstay of therapy for hemophilia is replacement of the deficient coagulation factor, through the use of purified concentrates either manufactured through recombinant technology or from pooled plasma. In the past, whole blood, plasma, or cryoprecipitate was used for replacement therapy. Factor concentrates are advantageous as they are generally accessible, easily handled and stored, virally inactivated, and commonly result in consistent hemostatic results. The dosage, frequency of administration, and duration of therapy depend on the activity level required, the half-life of the procoagulant, the intervention or procedure contemplated, or the location and severity of the bleeding episode. The half-life of factor VIII is approximately 12 hours, whereas for factor IX it is approximately 18 hours.10
Hemophilia A
Factor VIII concentrate is used for treatment of hemophilia A. Vials of factor concentrate are labeled with the number of international activity units contained, where 1 IU is the amount of activity of the procoagulant present in 1 mL of normal plasma. For routine hemorrhagic episodes, such as early joint, soft tissue, and oral bleeds, a one-time correction to approximately a level of 40% to 50% will achieve hemostasis and resolution of the bleeding episode. For mild factor VIII–deficient hemophilia, DDAVP (1-deamino-8-d-arginine vasopressin) (Sanofi-Aventis, Bridgewater, NJ) may be used for minor hemorrhagic episodes to achieve hemostasis. DDAVP (desmopressin acetate) is a synthetic analogue of the natural pituitary hormone 8-arginine vasopressin (antidiuretic hormone) affecting renal water conservation. This drug, when given intravenously, subcutaneously, or intranasally (Stimate) causes a rise in factor VIII activity and VWF through release from stored sites in endothelial cells, often to the hemostatic range. An appropriate rise in factor VIII activity to hemostatic levels should be documented for any given patient before therapeutic use of this agent, because response may vary among individuals. Peak levels are obtained approximately 1 hour after administration via intravenous and subcutaneous routes and 90 minutes after administration intranasally. The intranasal form of this medication, which is used to treat patients with bleeding disorders, has a more concentrated form of DDAVP compared with the preparation used to treat diabetes insipidus or enuresis. Therefore this preparation should be written for brand name only or in conjunction with the stated concentration of 1.5 mg/mL of desmopressin acetate. Repeated administration of DDAVP may result in tachyphylaxis, a reduction in expected response with sequential dosing due to depletion of storage sites. Use of DDAVP to treat hemorrhagic disorders may also be associated with water retention, hyponatremia, and rarely seizures; therefore monitoring of electrolytes may be required in some circumstances, especially in surgical situations.11–13
Hemophilia B
Factor IX–deficient hemophilia is treated with purified coagulation factor IX concentrate (monoclonal and recombinant). In the past, less pure products in the class of prothrombin complex concentrate (PCC) were used. PCCs contained other vitamin K–dependent coagulation factors in addition to factor IX, including some activated forms of these procoagulants. Individuals who require high doses or repeated infusions of PCC are at risk for development of disseminated intravascular coagulation and thrombosis. The minimal desired level for hemostasis is the same for factor IX as for factor VIII (40%). However, the number of units required to achieve that level is different as the volume of distribution of plasma-derived factor IX (1.0) is greater than that for factor VIII (0.5). The volume of distribution of recombinant factor IX is greater than that of plasma-derived factor IX (estimated minimum volume of 1.2 compared with 1, respectively, whereas in infants and young children a minimum volume of distribution of 1.4 should be used for dose calculation). Because interindividual variability of volume of distribution is wide, measurement of activity levels may be required to document a hemostatic level.14–17
Clotting factor concentrates are administered in different regimens depending on the patient’s level of severity, number of bleeding episodes, and the treating physician’s recommendations. Treatment regimens may be divided into replacement therapy administered after a bleeding episode has occurred (on-demand therapy) or as administered on a regular scheduled basis to prevent or suppress bleeding episodes (prophylaxis). Prophylactic regimens are further subdivided into primary and secondary. Primary prophylaxis is a long-term treatment for prevention of joint disease instituted before or after minimal hemarthrosis has occurred; secondary prophylaxis may be long or short term, but is instituted after hemarthrosis has occurred or to interrupt a bleeding pattern to rest a joint. Primary prophylactic therapy has been shown in a prospective randomized study to be the most effective regimen to prevent joint disease in patients with severe hemophilia and is now considered the standard of care for these patients.18 Therefore dental care providers should schedule dental evaluations and interventions on regularly planned infusion days, whereas patients treated with on-demand regimens require discussion regarding the need to administer replacement therapy specifically for dental interventions. Patients using regimens of prophylaxis may have a central venous catheter placed due to the need for frequent venous access. The use of antibiotic prophylaxis to protect the central venous access device may be considered although is not recommended by the Centers for Disease Control and Prevention (see Antibiotic Prophylaxis in the section on Dental Management).
WOMEN WITH BLEEDING DISORDERS
Von Willebrand Disease
Patients with von Willebrand disease should undergo subtyping to determine optimal therapy. DDAVP may be used to achieve hemostasis in most patients with type I von Willebrand disease, where type I VWD represents a quantitative VWF deficiency with intact multimers. When DDAVP is used, a test dose should be administered to document an adequate hemostatic response. For patients with less common subtypes of VWD, patients who do not respond to DDAVP, or patients for whom DDAVP is inappropriate, or in bleeding events for which DDAVP should not be used, other therapeutic modalities may be required, including replacement with exogenous intact VWF through the use of a concentrate. Interventions and therapeutic approaches should be discussed with a hemophilia-comprehensive treatment center.19,20
Female carriers of hemophilia A and B may have decreased levels of factors VIII and IX, respectively, that place them in the mild range of deficiency. It is recommended that all carriers of hemophilia have an evaluation to determine their baseline activity level specific to the type of hemophilia carried to determine bleeding risk. Women who are carriers of hemophilia should be treated as potential mild-deficient patients and their hematologist contacted to determine the baseline factor activity level and need for treatment before or after specific dental interventions.21,22
COMPLICATIONS
Inhibitors are antibodies that neutralize the replaced coagulation factor and are one of the most severe complications for patients. Inhibitors may develop in approximately 28% of patients with severe factor VIII deficiency and in 3% to 5% of patients with severe factor IX deficiency. The key to successful treatment of patients with inhibitors is accurate knowledge of the classification and level of the inhibitor. Patients with inhibitors are divided into two general groups, high responders and low responders, based on the past peak anamnestic response of the inhibitor titer. Inhibitor levels are measured in Bethesda units (BU), a measurement that reflects the ability of the antibody to neutralize a specific amount of procoagulant.23,24
Other complications of hemophilia include arthritis and degenerative joint disease secondary to recurrent bleeding.25 Blood-borne viral infections represent an important complication of treatment of these disorders and may have been transmitted via required blood or blood products. Hepatitis, including both B and C and resultant liver disease have been a significant source of morbidity and mortality in this patient population.26 The human immunodeficiency virus (HIV) has also been a major source of morbidity and mortality since approximately 1979. Before 1985, there was no antibody test for HIV and no consistent method of viral inactivation in the manufacture of factor concentrates. Therefore between 1979 and 1985, factor concentrates and blood products may have been contaminated with HIV. Approximately 90% of hemophilic patients with severe factor VIII deficiency and 30% of those with severe factor IX deficiency who received factor concentrate during the at-risk period may have become infected with HIV. HIV infection is a sensitive issue to these individuals, who may now bear the burden of two chronic conditions.27 Currently available treatments of factor concentrates made through recombinant technology or pooled plasma have effectively eliminated transmission of HIV and hepatitis B and C. Nevertheless, universal precautions should be followed when treating all hemophilic patients with a history of receiving either factor concentrate.
RISKS TO DENTAL STAFF
A study by Klein and colleagues demonstrated a less than 0.5% occupational risk for HIV infection among dental professionals despite their infrequent compliance with recommended infection control precautions, frequent occupational exposure to persons at increased risk for HIV infection, and frequent accidental parenteral inoculations with sharp instruments.28 These data are reassuring but do not obviate the need for appropriate universal precautions with all patients.
DEVELOPMENT OF A TREATMENT PLAN
The dentist must be fully aware of the procedures that can be safely performed and those in which complications may arise. The dentist should confer with the patient’s physician and hematologist to formulate an appropriate treatment plan. The dentist should know the specific type of bleeding disorder, the severity of the disorder, the frequency and treatment of bleeding episodes, and the patient’s inhibitor status. Many individuals with hemophilia self-administer infusion products at home and are therefore able to treat themselves when required. The dentist should be prepared to discuss with the hematologist the type of anesthetic anticipated to be administered, the invasiveness of the dental procedure, the amount of bleeding anticipated, and the time involved in oral wound healing to help establish an appropriate treatment plan including the need for replacement and adjunctive therapies.29
USE OF ANTIFIBRINOLYTIC AGENTS
 -aminocaproic acid (Amicar, Xanodyne Pharmaceuticals, Florence, KY) and tranexamic acid (Cyklokapron, Pfizer, New York). Hemophilic patients form loose, friable clots that may be readily dislodged or quickly dissolved, especially in the oral cavity where local fibrinolysis is increased. Antifibrinolytics prevent clot lysis within the oral cavity. They are often used as an adjunct to factor concentrate replacement. For some dental procedures in which minimal bleeding is anticipated, they may be used alone.
-aminocaproic acid (Amicar, Xanodyne Pharmaceuticals, Florence, KY) and tranexamic acid (Cyklokapron, Pfizer, New York). Hemophilic patients form loose, friable clots that may be readily dislodged or quickly dissolved, especially in the oral cavity where local fibrinolysis is increased. Antifibrinolytics prevent clot lysis within the oral cavity. They are often used as an adjunct to factor concentrate replacement. For some dental procedures in which minimal bleeding is anticipated, they may be used alone.Dosages
 -aminocaproic acid is given immediately before dental treatment in an initial loading dose of 100 to 200 mg/kg by mouth up to a maximum total dose of 10 g. Subsequently, 50 to 100 mg/kg per dose up to a total maximum dose of 5 g is administered orally every 6 hours for 5 to 7 days. Alternatively, for patients of approximately adult size or heavier than 30 kg, a regimen of 3 g by mouth four times daily without a loading dose may be used. The advantage of
-aminocaproic acid is given immediately before dental treatment in an initial loading dose of 100 to 200 mg/kg by mouth up to a maximum total dose of 10 g. Subsequently, 50 to 100 mg/kg per dose up to a total maximum dose of 5 g is administered orally every 6 hours for 5 to 7 days. Alternatively, for patients of approximately adult size or heavier than 30 kg, a regimen of 3 g by mouth four times daily without a loading dose may be used. The advantage of  -aminocaproic acid for children is that it is available in both tablet and liquid form.
-aminocaproic acid for children is that it is available in both tablet and liquid form.The adult and pediatric dosage of tranexamic acid is 25 mg/kg given immediately before dental treatment. The same dose is continued every 8 hours for 5 to 7 days. The oral preparation of tranexamic acid is not available in the United States but the intravenous formulation is. The intravenous formulation may be administered orally if required.30
 -aminocaproic acid or replacement therapy. However, if the infiltration injection is into loose connective tissue or a highly vascularized area, then factor concentrate replacement to achieve a level of approximately 30% to 40% activity is recommended.
-aminocaproic acid or replacement therapy. However, if the infiltration injection is into loose connective tissue or a highly vascularized area, then factor concentrate replacement to achieve a level of approximately 30% to 40% activity is recommended.DENTAL MANAGEMENT
Most hemophilic patients can receive outpatient dental care routinely. Appointments should be arranged so that maximum treatment is accomplished per visit to minimize the need for unscheduled factor infusions and hence cost. Patients with inhibitors are best treated at a center with experience in dealing with this complication.31–33
Prevention of Dental Disease
A program that includes toothbrushing, flossing, appropriate topical fluoride exposure, and adequate systemic fluoride administration, as well as consumption of a proper diet and professional examination at regular intervals are effective measures that prevent dental problems. Rubber cup prophylaxis and supragingival scaling may be safely performed without prior factor replacement therapy. Minor bleeding can be readily controlled with local measures, such as direct pressure with a moistened gauze square. If bleeding persists for several minutes, the topical application of bovine thrombin,* microfibrillar collagen (Avitene, Medchem Products, Inc., Woburn, MA), and local fibrin glue may be of value.
Periodontal Therapy
Patients who require deep scaling because of gross calculus should initially undergo supragingival scaling. The tissue should be allowed to heal for 7 to 14 days, during which time the gingiva recede as edema and hyperemia diminish. Subsequent treatments to remove calculus and irritants therefore incur decreased bleeding risk from the tissue. If subgingival scaling is planned, replacement therapy may be considered, depending on the amount of anticipated bleeding and the severity of the factor deficiency. It is imperative that periodontal patients be placed on a maintenance schedule for proper management.34,35
Restorative Procedures
High-speed vacuum and saliva ejectors must be used with caution so that sublingual hematomas do not occur. Care must also be used in the placement of intraoral radiographic films, particularly in highly vascular sublingual tissues.
Oral Surgery
Preoperative evaluation and postoperative management of the hemophilic patient undergoing extractions must be coordinated with the hematologist. The dentist should discuss with the hematologist the surgical procedure, including the anesthetic technique, the degree of anticipated surgical trauma, and the expected duration for healing. The hematologist can then determine the amount and duration of factor concentrate replacement and adjunctive therapies required for surgery and postoperative management. Today it is possible to perform oral surgery in the hemophilic patient on an outpatient basis.36,37 Requirements include an experienced dentist and hematologist, a facility available for the patient to receive infusions if home infusion is not performed, and a coagulation laboratory capable of timely needed laboratory evaluations. Patients with inhibitors should only be treated in a hospital setting by those experienced in their management.
Antibiotic Prophylaxis
Total joint replacement, usually of the hip or knee, is often performed in adult patients with severe hemophilia to restore function and alleviate pain associated with degenerative arthritis due to multiple hemarthroses. Antibiotic prophylaxis is required for patients with artificial joints before invasive dental procedures. The American Heart Association recommendations for bacterial endocarditis prophylaxis, last updated in 2007, are commonly followed. Antibiotic prophylaxis is no longer recommended for patients with central venous access devices, although each particular patient’s circumstance should be considered.38 If the patient is immunocompromised because of HIV infection, intravenous antibiotic prophylaxis may be considered.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


