CHAPTER 24 Antiarrhythmic Drugs
BASIC CARDIAC ELECTROPHYSIOLOGY
Under normal conditions, the chambers of the heart contract as synchronized rhythmic units driven by electrical impulses generated in and conducted throughout the heart. The normal pacemaker impulse is generated in the sinoatrial (SA) node and travels through the atria to each muscle cell, to the atrioventricular (AV) node, and through specialized conduction pathways in the common bundle of His, bundle branches, and Purkinje network to reach the ventricular muscle cells. Figure 24-1 illustrates representative action potentials for an SA nodal cell, an atrial muscle cell, an AV nodal cell, a Purkinje fiber, and a ventricular muscle cell. Three experimental measures are used to characterize the electrophysiologic properties of the heart: automaticity, refractoriness, and conduction velocity. Many of the antiarrhythmic effects of drugs result from changes in these parameters, which are reflected by action potential alterations in various regions of the heart.
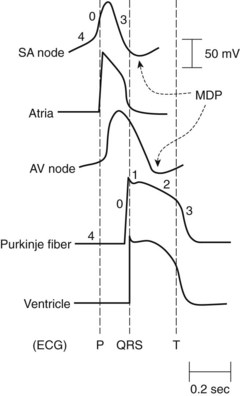
Automaticity
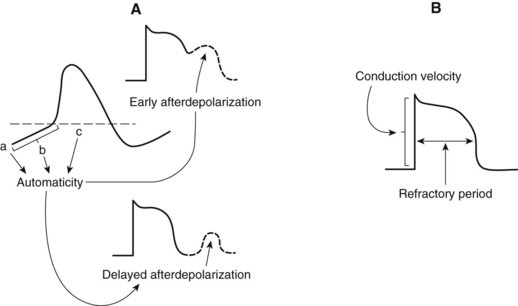
Automaticity describes the unique ability of cells of the SA node, AV node, and specialized conducting system to exhibit spontaneous phase 4 depolarization and impulse generation. An increase in automaticity refers to an increase in the rate of impulse generation, and, conversely, a decrease in automaticity refers to a decrease in the rate of impulse generation. Under normal conditions, the pacemaker cells of the SA node exhibit the most rapid generation of impulses, making the SA node the controlling pacemaker of the heart. The rate at which pacemaker cells initiate impulses is a function of the rate of phase 4 depolarization, the maximum diastolic potential (MDP), and the magnitude of the threshold potential (Figure 24-2). An increase in the rate of phase 4 depolarization in the SA node increases heart rate, whereas a change in the threshold voltage to a more positive value or an increase in the MDP (hyperpolarization) decreases the heart rate. These functions are under nervous and hormonal control and can be altered by injury or drugs.
Refractoriness
The period after the initiation of an action potential during which another action potential cannot be initiated and propagated regardless of stimulus is known as the effective refractory period (ERP) (see Figure 24-2). A change in the action potential duration (APD) is accompanied by a similar change in the duration of the ERP, although the ratio of change may not be 1 : 1. If the ERP is lengthened with respect to the APD, the cardiac cells will have repolarized more completely before they respond to a stimulus. Many drugs with antiarrhythmic effects prolong the duration of the ERP, and some decrease it.
Ion Channels
Ions and the channels that control their movements play major roles in the various phases of cardiac depolarization and repolarization. Figure 24-3 illustrates the membrane action potential in an SA nodal cell and a Purkinje fiber—two characteristically different action potentials—and the flow of ions through specific channels in the Purkinje fiber.
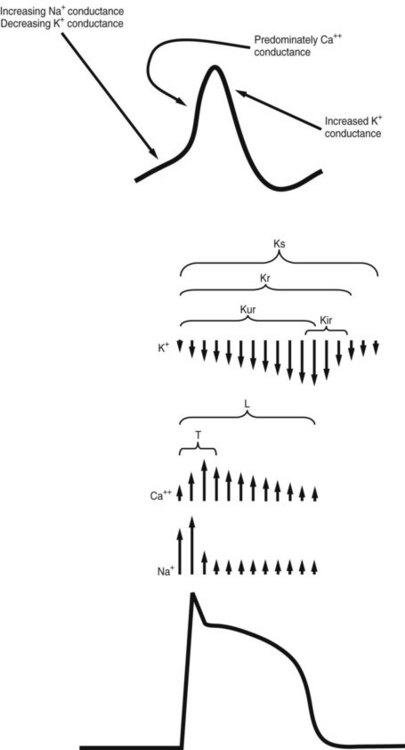
The K+ current that is responsible for repolarization of the action potential is termed the delayed outwardly rectifying K+ current (IK). IK is composed of several distinct currents carried through separate channels. Each current and its corresponding channel are defined by the rapidity with which they activate. The K+ currents IKs, IKr, and IKur, referring to slow-activating, rapid-activating, and ultrarapid-activating currents, are conducted through Ks, Kr, and Kur channels.13
ORIGINS OF ARRHYTHMIAS
Rhythm disturbances, often occurring as a result of myocardial infarction, are the most common cause of death from heart disease. Arrhythmias are thought to originate from abnormal impulse generation, impulse conduction, or both in combination. Some arrhythmias caused by abnormal impulse generation result from increased automaticity. These tachyarrhythmias are usually in response to an increase in the rate of diastolic depolarization (increased slope of phase 4) in pacemaker cells. Phase 4 depolarization can be altered by autonomic nervous system activity, by hormones, or by drugs. Changes in the MDP and threshold potential voltage can also affect automaticity. Abnormal impulse generation may also be triggered by afterpotentials that occur in cardiac pacemakers affected by drugs, disease, or other disturbances (see Figure 24-2). The induced afterdepolarizations may be early (before repolarization is complete) or delayed (after full repolarization has occurred) and can result in sustained tachyarrhythmias.1 Excessive intracellular Ca++ is a major contributor to delayed afterpotentials, whereas delayed repolarization increases the risk for early afterdepolarizations in some cells.
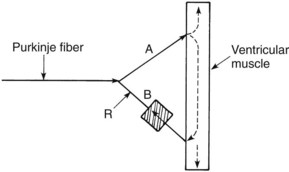
An important example of an alteration in impulse conduction that is easily induced in experimental animals is the phenomenon known as reentry. Figure 24-4 shows how a reentrant rhythm may develop. As illustrated, conduction in branch A is normal, whereas impulses in branch B can proceed in only the reverse direction (unidirectional block). A normally conducted impulse through branch A can be conducted in retrograde fashion through branch B to re-excite an area of tissue (point R) that was previously excited by the normal path of conduction. For this “circus movement” to occur, the tissue at point R must have repolarized to a point at which excitation is possible (which usually means that the retrograde conduction is relatively slow). A wave of re-excitation traveling in a circular path through fiber A, the contractile cardiac muscle, and fiber B can result in a self-sustaining arrhythmia. Reentry is usually a major contributor to atrial fibrillation, an arrhythmia especially common in elderly individuals.
Disturbances in the relationship of the fast and slow electrical responses of certain cardiac cells may play an important role in the genesis of arrhythmias. The fast response refers to the rapid phase 0 depolarization caused by rapid Na+ influx (see Figure 24-3). This kind of activity is seen in atrial and ventricular muscle fibers and specialized conducting fibers. In addition to the rapid inward current carried by Na+, the fast fibers exhibit a second, slower inward current carried by Ca++. The slower current does not normally constitute a major factor in phase 0 depolarization of the atrial and ventricular myocardium and Purkinje fibers, but it persists after rapid depolarization and is responsible for the prolonged plateau phase characteristic of these fibers. Fibers located in the SA and AV nodes, the AV ring fibers, and the mitral and tricuspid valve leaflets show the slow response in phase 0, during which the depolarization is carried largely by the inward Ca++ current.
Certain arrhythmias can be traced to defects in one or more ion channels. The long QT syndrome results from delayed repolarization in the ventricle. A delayed repolarization can be caused by any depolarizing current, such as a Na+ current, that lingers into phase 3 of the action potential. It can also result from reduced activity of a repolarizing K+ current. A defect in the Kr (rapid activating current) channel is the basis for one type of familial long QT syndrome that can devolve into torsades de pointes, a potentially life-threatening ventricular tachyarrhythmia (see later).10 Torsades de pointes may also be elicited by drugs that inhibit Kr channels and increase the QT interval. These include numerous antiarrhythmic agents and some drugs of other classes. Whether the delay in repolarization is caused by a hereditary defect or by a drug, it leads to a net enhancement of inward cationic flow, which can trigger early afterdepolarizations (see Figure 24-2). Because the cells in the wall of the ventricle are not equally affected, multiple waves of reentry can occur, initiating torsades de pointes. Effort is under way to develop more selective K+ channel inhibitors as potential antiarrhythmic drugs. Torsades de pointes is a major risk of drugs that selectively block Kr channels. Hypokalemia and hypomagnesemia increase the risk of developing torsades de pointes.16
ELETROCARDIOGRAPHY AND COMMON ARRHYTHMIAS
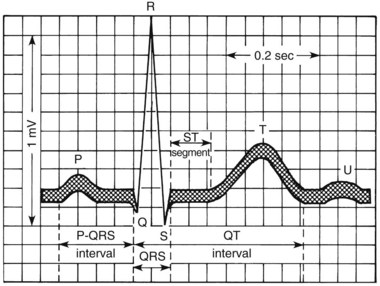
Arrhythmias are generally classified as supraventricular (originating in the atria or conducting system not in the ventricle) or ventricular. A few of the most common arrhythmias are described. For comparison, a diagram of a normal electrocardiogram (ECG) is provided in Figure 24-5. In Figure 24-5, also note the P–QRS (or PR), QT, and ST intervals and the duration of the QRS complex. Figure 24-6 contains representations of ECGs recorded during arrhythmias of ventricular and supraventricular origin.
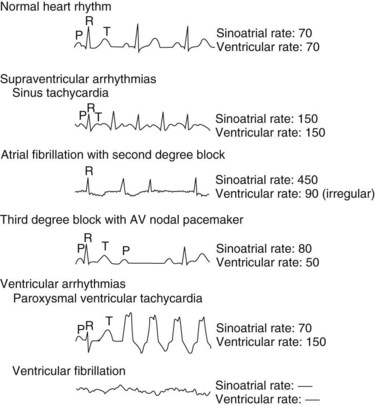
The first arrhythmia illustrated is a simple sinus tachycardia caused by rapid impulse generation (i.e., increased automaticity) in the SA node. Higher rates of atrial activity often involve reentry, as in atrial flutter (approximately 300 beats/min) or fibrillation (400 to 700 beats/min).7 Under these conditions, second-degree heart block occurs, as characterized by the failure of some atrial depolarizations to initiate a QRS complex. In a third-degree block (also shown), there is complete dissociation between atrial and ventricular contractions. The ventricular arrhythmias are caused by the development of ectopic foci or reentrant conduction in the ventricles. The first one shown in Figure 24-6 is ventricular tachycardia. In ventricular fibrillation, the most immediately life-threatening arrhythmia, erratic depolarization of different areas of the ventricle totally disorganizes myocardial contraction, renders the heart ineffective, and causes the cardiac output to plummet. Immediate treatment of ventricular fibrillation, usually including defibrillation (precordial direct current shock), must be provided to avert sudden death.
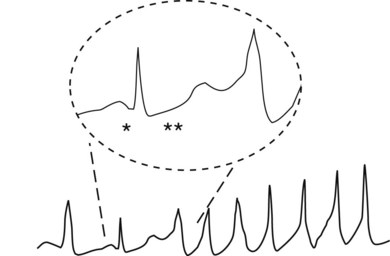
Torsades de pointes (literally meaning “twisting of points”) is a polymorphic ventricular tachycardia characterized by bizarre shapes in the ventricular depolarization complexes on the ECG (Figure 24-7). As mentioned previously, it often occurs in patients with defective K+ channel (e.g., Kr) activity and occurs with certain drugs that delay repolarization of ventricular muscle cells, often by blocking Kr channels. In both cases, QT prolongation precedes and leads to torsades de pointes. In Figure 24-7, an excessively long QT interval is followed by a ventricular tachycardia in which each depolarization has a different configuration.
ANTIARRHYTHMIC DRUGS
Drugs used in the treatment of cardiac arrhythmias are not easily classified because they often have more than one action. Drugs within each class vary in their magnitudes of action or types of effects produced.1,14 The most common scheme, originally proposed by Vaughan and Williams,14 classifies drugs according to certain specific properties. Type I drugs, such as quinidine, lidocaine, and flecainide, depress Na+ current.1 The type I agents are subdivided further according to their relative effects on phase 0 depolarization, conduction velocity, and APD. Na+ channels exist in at least three states: closed, open, and inactivated. At resting membrane potentials the Na+ channels are closed except for a Na+ “leak” associated with phase 4 depolarization for cells that display automaticity. During rapid depolarization (phase 0, especially in Purkinje fibers and ventricular muscle), the Na+ channels are open. The Na+ channels convert to the inactivated state before returning to the resting, closed state. The inactivated state occurs mostly in phase 2 and 3 of the action potential.
Quinidine-like, or class IA, drugs depress phase 0 depolarization at all heart rates. They prolong the APD of the ventricle because they also inhibit K+ (chiefly Kr) channels (Figure 24-8). Class IB agents, such as lidocaine, block Na+ channels more selectively, but the rapid onset and recovery of Na+ channel blockade results in little accumulated lidocaine effect on phase 0 and conduction velocity in healthy tissue at normal heart rates. In damaged or rapidly firing cells, lidocaine causes a frequency-dependent or use-dependent block to reduce the slope of phase 0 and lowers phase 4 in ectopic pacemakers and in Purkinje fibers under high sympathetic tone.1 The faster the heart rate, the greater is the effect of lidocaine. (Use-dependent block is discussed in Chapter 16.) In contrast to other class I agents, lidocaine and related class IB antiarrhythmics may actually shorten the APD. Flecainide and other class IC antiarrhythmics are characterized by their profound depression of phase 0 depolarization and slowing of conduction in the atria, AV node, and ventricles at normal heart rates. This pronounced effect results from their slow dissociation from Na+ channels and accumulation of the channel-blocking effect over several contraction cycles. There is little or no prolongation of the APD.
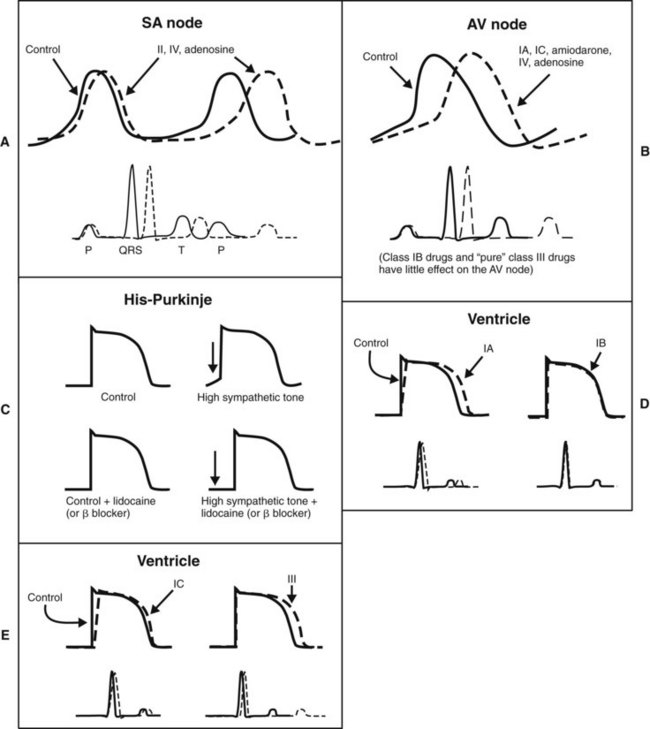
Propranolol and related β-adrenergic–blocking agents constitute class II drugs and inhibit cardiac stimulation brought on by β-adrenergic agonists. They depress phase 4 depolarization (see Figure 24-8). The class III group, including amiodarone and sotalol, block K+ channels (chiefly Kr channels) and prolong the APD by delaying phase 3 repolarization. Verapamil and other class IV drugs selectively block Ca++ channels (L type) and depress slow fiber conduction (phase 0 of the SA and AV nodes) and phase 4 depolarization (see Figure 24-8). Drugs that cannot be classified by the Vaughan-Williams scheme include digitalis and adenosine.
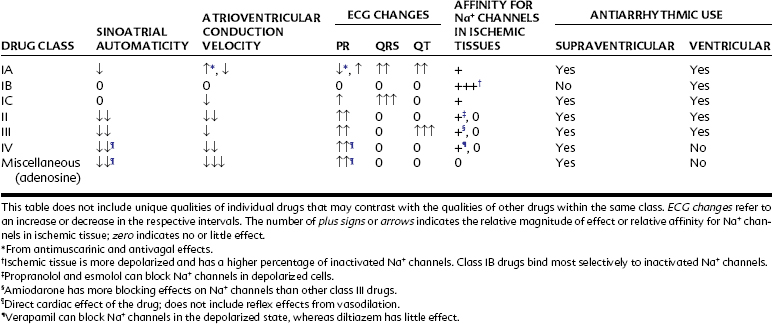
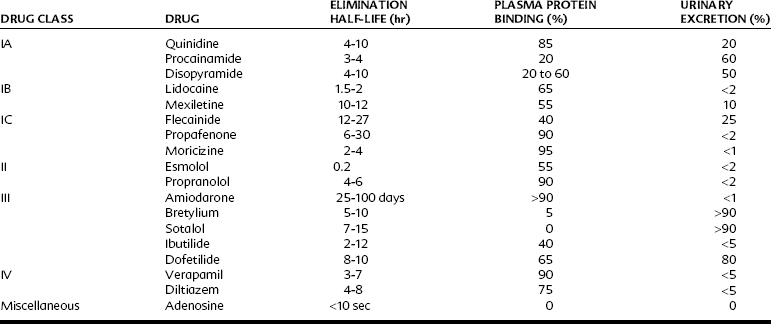
Table 24-1 outlines the various categories of antiarrhythmic agents. The drugs vary widely in their clinical usefulness. Class IA drugs are less commonly used today, partly because of the introduction of class IC and class III drugs. In Table 24-1 the action responsible for the classification of each drug, which is usually its major action, is circled. In the discussion of individual agents that follows, reference should also be made to Table 24-2 for the electrophysiologic actions of representative antiarrhythmic drugs. The net effects of the relevant drug classes and various action potentials in the heart are shown in Figure 24-8. Pharmacokinetic data for specific drugs are given in Table 24-3. The use of digoxin for certain kinds of arrhythmias is discussed in Chapter 25.

Quinidine
Quinidine is effective in the treatment of some atrial and (to a lesser extent) ventricular tachyarrhythmias. It was used clinically before its antiarrhythmic properties were discovered. During treatment with quinine and quinidine for patients with malaria, the reversal of atrial fibrillation was noted in some patients. Widespread use of quinidine for supraventricular arrhythmias followed reports from Wenckebach15 in 1914.
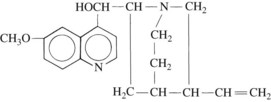
Quinidine, the d isomer of quinine, is found in the bark of the cinchona tree, which is indigenous to certain regions of South America. Synthesis of this compound has been accomplished, but the synthesized drug is expensive, and quinidine is still isolated from the natural source. Its structural formula is shown in Figure 24-9.
Pharmacologic effects
Quinidine reduces automaticity and conduction velocity and increases refractoriness. Automaticity is depressed through an increase in the threshold potential and a decrease in the slope of spontaneous diastolic depolarization (phase 4) in pacemaker fibers, particularly at sites other than the SA node. Quinidine has the potential to slow or abolish tachyarrhythmias. Quinidine decreases the slope of phase 0 depolarization and decreases conduction velocity in cells such as those of the AV node and ventricular myocardium (see Figure 24-8). By this effect, quinidine may inhibit reentrant pathways. Quinidine influences automaticity and conduction velocity by blocking Na+ channels, particularly channels in the open state. The rate of recovery from quinidine block is intermediate between class IB an/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


