Chapter 24
Acquired Bleeding and Hypercoagulable Disorders
A number of procedures that are performed in dentistry may cause bleeding. Under normal circumstances, these procedures can be performed with little clinical risk; however, in the patient whose ability to control bleeding has been altered by drugs or disease such procedures may be associated with potentially catastrophic outcomes unless the dental practitioner identifies the problem before initiation of treatment. In most instances, once the patient with a bleeding problem due to drugs or disease has been identified, appropriate dental management will greatly reduce the associated risks. This chapter presents an overview of the physiologic mechanisms involved in the control of bleeding and the pathophysiology of acquired bleeding disorders and hypercoagulable states. Congenital bleeding disorders and genetic hypercoagulable conditions are covered in < ?xml:namespace prefix = "mbp" />
Definition
Bleeding disorders are conditions that alter the ability of blood vessels, platelets, and coagulation factors to maintain hemostasis. Acquired bleeding disorders may occur as the result of diseases, drugs, radiation, or chemotherapy for cancer in which vascular wall integrity, platelet production or function, or coagulation factors are impaired.
Most bleeding disorders are iatrogenic. Every patient who receives coumarin to prevent recurrent thrombosis has a potential bleeding problem. Most of these patients are receiving anticoagulant medication because they have had a recent myocardial infarction, a cerebrovascular accident, or thrombophlebitis. Patients who have atrial fibrillation; who have had open heart surgery to correct a congenital defect, replace diseased arteries, or repair or replace damaged heart valves; or who have had recent total hip or knee replacement also may be receiving long-term anticoagulation therapy. Patients treated with antiplatelet medications to prevent cardiovascular complications also may have a potential bleeding problem. Some people treated with aspirin for chronic illnesses, such as rheumatoid arthritis, may have potential bleeding problems.
In a dental practice of 2000 adults, about 100 to 150 patients may have a possible bleeding problem. This is a rough estimate, however, and the number could be higher.
Epidemiology: Incidence and Prevalence
Patients on low-intensity warfarin therapy (with an international normalized ratio [INR] goal of 2.0 to 3.0) for prophylaxis of venous thromboembolism have a risk of major bleeding of less than 1% and about an 8% risk for minor bleeding. Patients on high-intensity warfarin therapy (with an INR goal of 2.5 to 3.5) have up to a five-fold greater risk for bleeding.
Patients with acute or chronic leukemia may have clinical bleeding tendencies because of thrombocytopenia, which may result from overgrowth of malignant cells in the bone marrow that leaves no room for red blood cells or platelet precursors. In addition, leukemic patients may develop thrombocytopenia from the toxic effects of the various chemotherapeutic agents used to treat the disease. The etiology and incidence of leukemia are reviewed in
It is difficult to obtain accurate information about the incidence of other systemic conditions, such as liver disease, renal failure, thrombocytopenia, and drug-induced vascular wall defects, that may render the patient susceptible to prolonged bleeding after injury or surgery. However, when the prevalence of drug-influenced or disease-produced defects in the normal control of blood loss is considered, a busy dental practice will contain a large number of patients who may be potential “bleeders.”
Etiology
A pathologic alteration of blood vessel walls, a significant reduction in the number of platelets, defective platelets or platelet function, a deficiency of one or more coagulation factors, the administration of anticoagulant or antiplatelet drugs, a disorder of platelet release, or the inability to destroy free plasmin can result in significant abnormal clinical bleeding. This may occur even after minor injuries and may lead to death in some patients if immediate action is not taken.
The classification given in

Box 24-1 Classification of Acquired Bleeding and Thrombotic Disorders

Infections, chemicals, collagen disorders, or certain types of allergy can alter the structure and function of the vascular wall to the point that the patient may have a clinical bleeding problem. A patient may have normal numbers of platelets, but they may be defective or unable to perform their proper function in the control of blood loss from damaged tissues. If the total number of circulating platelets is reduced to below 50,000/µL of blood, the patient may be a bleeder. In some cases, the total platelet count is reduced by unknown mechanisms; this is called primary or idiopathic thrombocytopenia. Chemicals, radiation, and various systemic diseases (e.g., leukemia) may have a direct effect on the bone marrow, potentially resulting in secondary thrombocytopenia.
Acquired coagulation disorders are the most common cause of prolonged bleeding. Liver disease and disseminated intravascular coagulation (DIC) can lead to severe bleeding problems. Many of the other acquired coagulation disorders may become apparent in patients only after trauma or surgical procedures. In contrast with the congenital coagulation disorders, in which only one factor is affected, the acquired coagulation disorders usually have multiple factor deficiencies.
The liver produces all of the protein coagulation factors; thus, any patient with significant liver disease may have a bleeding problem. In addition to having a possible disorder in coagulation, the patient with liver disease who develops portal hypertension and hypersplenism may be thrombocytopenic as a result of splenic overactivity, which leads to increased sequestration of platelets in the spleen.
Any condition that so disrupts the intestinal flora that vitamin K is not produced in sufficient amounts will result in a decreased plasma level of the vitamin K–dependent coagulation factors. Vitamin K is needed by the liver to produce prothrombin (factor II) and factors VII, IX, and X. Biliary tract obstruction, malabsorption syndrome, and excessive use of broad-spectrum antibiotics all can lead to low levels of prothrombin and factors VII, IX, and X on this basis.
Drugs, such as heparin and coumarin derivatives, can cause a bleeding disorder because they may disrupt the coagulation process. Antiplatelet medications, aspirin, other nonsteroidal antiinflammatory drugs (NSAIDs), penicillin, cephalosporins, and alcohol also may interfere with platelet function.
Many herbal supplements can impair hemostatic function for the control of bleeding. Fish oil or concentrated omega-3 fatty acid supplements may impair platelet activation. Diets naturally rich in omega-3 fatty acids can result in a prolonged bleeding time and abnormal platelet aggregation.
Pathophysiology
The three phases of hemostasis for controlling bleeding are vascular, platelet, and coagulation. The vascular and platelet phases are referred to as primary, and the coagulation phase is secondary. The coagulation phase is followed by the fibrinolytic phase, during which the clot is dissolved (

Box 24-2 Normal Control of Bleeding

Vascular Phase
The vascular phase begins immediately after injury and involves vasoconstriction of arteries and veins in the area of injury, retraction of arteries that have been cut, and buildup of extravascular pressure by blood loss from cut vessels. This pressure aids in collapsing the adjacent capillaries and veins in the area of injury. Vascular wall integrity is important for maintaining the fluidity of blood. The smooth endothelial lining consists of a nonwettable surface that, under normal conditions, does not activate platelet adhesion or coagulation. In fact, the endothelial cells synthesize and secrete three potent antiplatelet agents: prostacyclin, nitric oxide, and certain adenine nucleotides.
Vascular endothelial cells also are involved in antithrombotic and prothrombotic activities. The major antithrombotic activity consists of secretion of heparin-like glycosaminoglycans (heparin sulfate) that catalyze inactivation of serine proteases such as thrombin and factor Xa by antithrombin III. Endothelial cells also produce thrombomodulin, which combines with thrombin to form a complex that activates protein C. Activated protein C (APC) then binds to endothelially released protein S, causing proteolysis of factor Va and factor VIIIa that inhibits coagulation. Tissue-type plasminogen activator (tPA) is released by injured endothelial cells to initiate fibrinolysis.
Vessel wall components contribute prothrombotic activities. Exposure of vessel wall subendothelial tissues, collagen, and basement membrane through chemical or traumatic injury serves as a tissue factor (TF)—for which the old term was tissue thromboplastin—and initiates coagulation by way of the extrinsic pathway. The extrinsic pathway can be turned off by tissue factor pathway inhibitor (TFPI). An inducible endothelial cell prothrombin activator may directly generate thrombin. Injured endothelial cells release adenosine diphosphate (ADP), which induces platelet adhesion. Vessel wall injury also promotes platelet adhesion and thrombus formation through exposure of subendothelial tissues to von Willebrand factor (vWF). Endothelial cells also contribute to normal homeostasis and vascular integrity through synthesis of type IV collagen, fibronectin, and vWF.
Platelet Phase
Platelets are cellular fragments from the cytoplasm of megakaryocytes that last 8 to 12 days in the circulation. About 30% of platelets are sequestered in the microvasculature or spleen and serve as a functional reserve. Platelets do not have a nucleus; thus, they are unable to repair inhibited enzyme systems through drugs such as aspirin. Aged or nonviable platelets are removed and destroyed by the spleen and liver.
Subendothelial tissues are exposed at the site of injury and, through contact activation, cause the platelets to become sticky and adhere to subendothelial tissues, platelet membrane glycoprotein Ib (GPIb) binds with vWF, which is attached to the subendothelial tissue, and glycoprotein Ia/IIa (GPIa/IIa) and glycoprotein VI (GPVI) bind to collagen in the injured vessel wall.
ADP released by damaged endothelial cells initiates aggregation of platelets (primary wave), and when platelets release their secretions, a second wave of aggregation results. Platelets bind with fibrinogen by the membrane glycoprotein IIb (GPIIb) the fibrinogen is then converted to fibrin, which stabilizes the platelet plug. The result of the preceding processes is a clot of platelets and fibrin attached to the subendothelial tissue.

Box 24-3
Platelet Functions and Activation
2 Platelets contain three types of secretory granules:
3 Platelets provide a surface for activation of soluble coagulation factors:
Data from McMillan R: Hemorrhagic disorders: abnormalities of platelet and vascular function. In Goldman L, Ausiello D, editors: Cecil medicine, ed 23, Philadelphia, 2008, Saunders; and Baz R, Mekhail T: Disorders of platelet function and number. In Carey WD, et al, editors: Current clinical medicine 2009—Cleveland Clinic, Philadelphia, 2009, Saunders.

A product of platelets, thromboxane, is needed to induce platelet aggregation. The enzyme cyclooxygenase is essential in the process for generation of thromboxane. Endothelial cells, through a similar process (also dependent on cyclooxygenase), generate prostacyclin, which inhibits platelet aggregation. Aspirin acts as an inhibitor of cyclooxygenase, and this causes irreversible damage to the platelets. However, endothelial cells can, after a short period, recover and synthesize cyclooxygenase; thus, aspirin has only a short effect on the availability of prostacyclin from these cells. The net result of aspirin therapy is to inhibit platelet aggregation. This effect can last up to 9 days (time needed for all old platelets to be cleared from the blood).
Coagulation Phase
The process of the fibrin-forming (coagulation) system is shown in
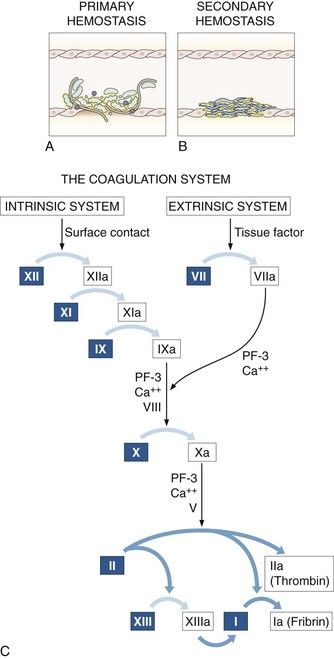
FIGURE 24-1 The primary (vascular/platelet) system (A), the secondary (coagulation) system for the control of bleeding (B), and the coagulation cascade (C). The intrinsic coagulation system is triggered by surface contact; the extrinsic system, by release of tissue factor from injured tissues; and the common pathway, by factor X.
(From Ragni MV: The hemophilias: factor VIII and factor IX deficiencies. In Young NS, Gerson SL, High KA, editors: Clinical hematology, St. Louis, 2006, Mosby.)
FIGURE 24-3 A colored scanning electron micrograph of a blood clot or thrombus inside the coronary artery of a human heart.
(Reprinted with permission of P. M. Motta, G. Macchiarelli, S. A. Nottola/Photo Researchers, Inc.)
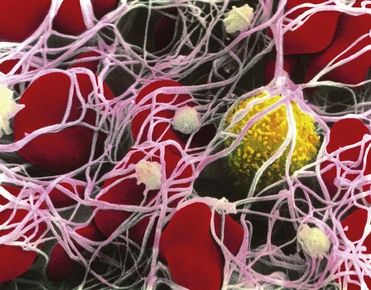
FIGURE 24-2 The end product of the coagulation system, which shows a fibrin clot or thrombus. White threads are fibrin, the structure with yellow on the surface is a white blood cell, platelets are green, and the red structures are red blood cells.
(Reprinted with permission of CNRI/Photo Researchers, Inc.)
Coagulation of blood involves the components shown in
TABLE 24-1 Blood Coagulation Components
| Factor | Deficiency | Function |
|---|---|---|
| Factor II (prothrombin) | Acquired—common | Protease zymogen |
| Factor X | Acquired—common | Protease zymogen |
| Factor IX | Acquired—common | Protease zymogen |
| Factor VII | Acquired—common | Protease zymogen |
| Factor VIII | Acquired—rare | Cofactor |
| Factor V | Acquired—rare | Cofactor |
| Factor XI | Acquired—common | Protease zymogen |
| Factor I (fibrinogen) | Acquired—common | Structural |
| von Willebrand Factor | Acquired—rare | Adhesion |
From McVey JH: Coagulation factors. In Young NS, Gerson SL, High KA, editors: Clinical hematology, St. Louis, 2006, Elsevier.
The (faster) extrinsic pathway is initiated through tissue factor (an integral membrane protein) and is released or exposed through injury to tissues; this process activates factor VII (VIIa). In the past, the trigger for initiating the extrinsic pathway was referred to as a tissue thromboplastin. It has since been shown that the real activator is the tissue factor (TF). The term extrinsic pathway continues to be used today, despite the fact that it is somewhat outdated. This is because TF is not always extrinsic to the circulatory system but is expressed on the surface of vascular endothelial cells and leukocytes.
Thrombin generated by the faster extrinsic and common pathway is used to accelerate the slower intrinsic and common pathway. Activation of factor XII acts as a common link between the component parts of the homeostatic mechanism: coagulation, fibrinolytic, kinin, and complement systems. As a result, thrombin is generated; in turn, fibrinogen is converted to fibrin, activates factor XIII, enhances factor V and factor VIII activity, and stimulates aggregation of additional platelets.
Fibrinolytic Phase
The fibrin-lysing (fibrinolytic) system is needed to prevent coagulation of intravascular blood away from the site of injury and to dissolve the clot, once it has served its function in homeostasis (
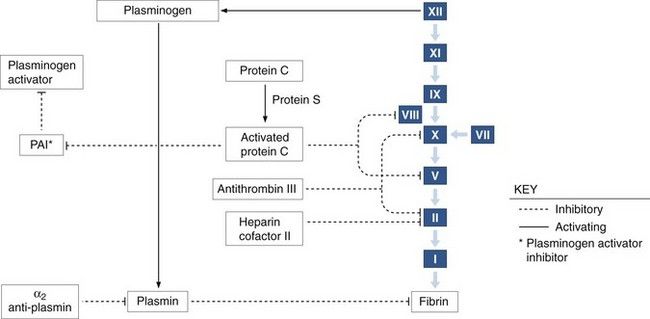
FIGURE 24-4 The coagulation and fibrinolytic pathways with inhibitors.
(From Bontempo FA: Hematologic abnormalities in liver disease. In Young NS, Gerson SL, High KA, editors: Clinical hematology, St. Louis, 2006, Mosby.)
The tPA released by injured endothelial cells binds to fibrin as it activates the conversion of fibrin-bound plasminogen to plasmin. Circulating plasminogen (i.e., not fibrin bound) is not activated by tPA. Thus, tPA is efficient in dissolving a clot without causing systemic fibrinolysis.
The effect of plasmin on fibrin and fibrinogen is to split off large pieces that are broken up into smaller and smaller segments. The final smaller pieces are called split products. These split products also are referred to as fibrin degradation products (FDPs). FDPs increase vascular permeability and interfere with thrombin-induced fibrin formation; this can provide the basis for clinical bleeding problems.

Box 24-4
Fibrin-Lysing (Fibrinolytic) System
1 Activation of coagulation also activates fibrinolysis.
3 Plasminogen activated to plasmin
Data from Lijnen HR, Collen D: Molecular and cellular basis of fibrinolysis. In Hoffman R, et al, editors: Hematology: basic principles and practice, Philadelphia, 2009, Churchill Livingstone; and Kessler CM: Hemorrhagic disorders: coagulation factor deficiencies. In Goldman L, Ausiello D, editors: Cecil textbook of medicine, ed 23, Philadelphia, 2008, Saunders.

Antiplasmin factors present in circulating blood rapidly destroy free plasmin but are relatively ineffective against plasmin that is bound to fibrin (

Box 24-5
Physiologic Antithrombotic Systems
1 Normal endothelium promotes blood fluidity by inhibiting platelet activation.
2 Endothelium also plays a role in anticoagulation by preventing fibrin formation.
4 Activated protein C, with its cofactor protein S, acts as a natural anticoagulant by destroying factors Va and VIIIa.
5 Tissue factor pathway inhibitor (TFPI), a plasma protease inhibitor, inhibits factor VIIa and the extrinsic pathway.
6 The endogenous fibrinolytic system degrades any fibrin produced despite the above-mentioned antithrombotic mechanisms.
7 Inherited deficiencies of antithrombin, protein C, or protein S are associated with a lifelong thrombotic tendency.
8 TFPI deficiency has yet to be related to clinical problems.
Data from Dahlback B, Stenflo J: Regulatory mechanisms in hemostasis: natural anticoagulants. In Hoffman R, et al, editors: Hematology: basic principles and practice, ed 5, Philadelphia, 2009, Churchill Livingstone.

Timing of Clinical Bleeding
A significant disorder that may occur in the vascular or platelet phase leads to an immediate clinical bleeding problem after injury or surgery. These phases are concerned with controlling blood loss immediately after an injury and, if defective, will lead to an early problem. However, if the vascular and platelet phases are normal, and the coagulation phase is abnormal, the bleeding problem will not be detected until several hours or longer after the injury or surgical procedure. In the case of small cuts, for example, little bleeding would occur until several hours after the injury, and then a slow trickle of bleeding would start. If the coagulation defect were severe, this slow loss of blood could continue for days. Even with this “trivial” rate, a significant loss of blood might occur (0.5 mL/minute or about 3 U/day).
Clinical Presentation
Signs and Symptoms
Signs associated with bleeding disorders may appear in the skin or mucous membranes or after trauma or invasive procedures. Jaundice (
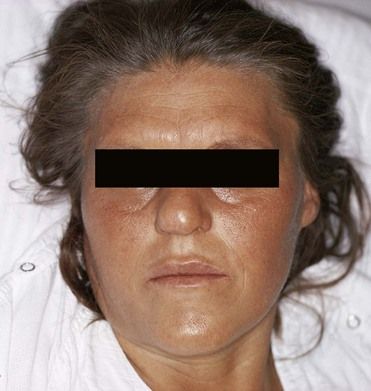
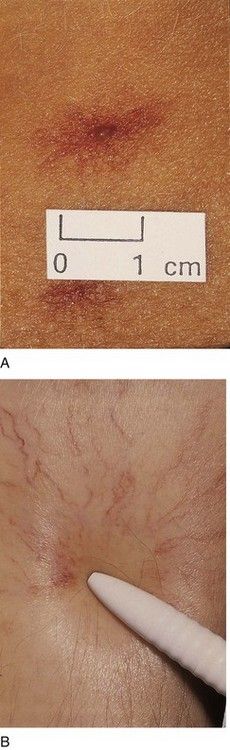
FIGURE 24-6 A, Spider angioma on the skin of a patient with chronic liver disease B, Note how the spider legs of the angioma blanch with pressure on the central arteriole.
(From Forbes CD, Jackson WF: Color atlas and text of clinical medicine, ed 3, Edinburgh, 2003, Mosby.)
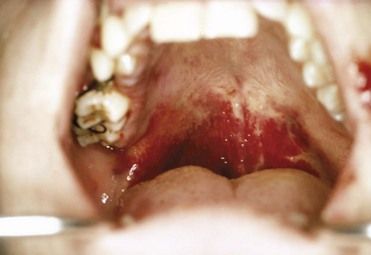
FIGURE 24-7 Ecchymoses on the mucosa of the hard and soft palate in a patient with chronic liver disease.
The signs seen most commonly in patients with abnormal platelets or thrombocytopenia are petechiae (
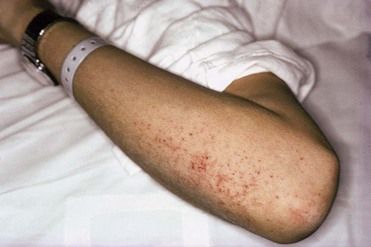
Patients with acute or chronic leukemia may reveal one or more of the following signs: ulceration of the oral mucosa, hyperplasia of the gingivae (
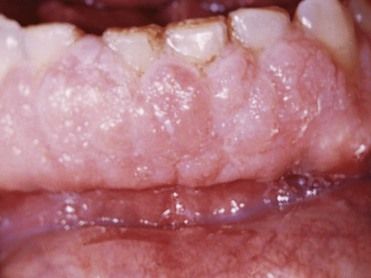
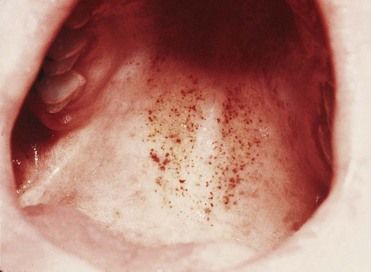
FIGURE 24-10 Palatal petechiae in a patient with leukemia.
(From Hoffbrand AV: Color atlas of clinical hematology, ed 3, St. Louis, 2000, Mosby.)
A number of patients with bleeding disorders may show no objective signs that suggest the underlying problem. Severe or chronic bleeding can lead to anemia with features of pallor, fatigue, and so forth. Anemia is discussed in detail in
Laboratory Tests
Several tests are available to screen patients for bleeding disorders and to help pinpoint the specific deficiency. In general, screening is done in dentistry when the patient reveals a history of a bleeding problem or a family member with a history of a bleeding problem, or when signs of bleeding disorders are found during the clinical examination. The dentist can order the screening tests, or the patient can be referred to a hematologist for screening. In medicine, routine screening is done for patients before major surgical procedures such as open heart surgery are performed.
The Ivy bleeding time (BT) has been used to screen for disorders of platelet function and thrombocytopenia. It has been found to be unreliable and is no longer used as a screening test. The platelet function analyzer (PFA-100), an instrument that measures platelet-dependent coagulation under flow conditions, is more sensitive and specific for platelet disorders and von Willebrand disease (vWD) than the bleeding time; however, it is not sensitive enough to rule out underlying mild bleeding disorders. Also, it has not been evaluated prospectively to determine its utility in predicting bleeding risk, although such studies are underway. Therefore, the BT and PFA-100 are not recommended as screening tests to be used by the dentist.
Three tests are recommended for use in initial screening for possible bleeding disorders:
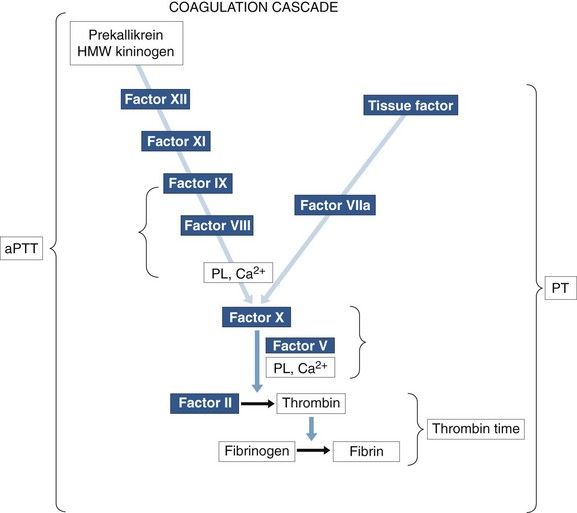
FIGURE 24-11 Coagulation cascade indicating the intrinsic pathway measured by activated partial thromboplastin time (aPTT), the extrinsic pathway measured by prothrombin time (PT), and the conversion of fibrinogen to fibrin, which is measured by thrombin time (TT). Other proteins—prekallikrein and high-molecular-weight (HMW) kininogen—participate in the contact activation phase but are not considered coagulation factors. Ca2+, Calcium; PL, Phospholipid.
(From Rick ME: Coagulation testing. In Young NS, Gerson SL, High KA, editors: Clinical hematology, St. Louis, 2006, Mosby.)
Patients with positive screening tests should be evaluated further so the specific deficiency can be identified and the presence of inhibitors ruled out. A hematologist orders these tests, establishes a diagnosis that is based on the additional testing, and makes recommendations for treatment of the patient who is found to have a significant bleeding problem.
Screening Tests:
Partial Thromboplastin Time
Partial thromboplastin time (PTT) is used to check the intrinsic system (factors VIII, IX, XI, and XII) and the common pathways (factors V and X, prothrombin, and fibrinogen). It also is the best single screening test for coagulation disorders. A phospholipid platelet substitute is added to the patient’s blood to initiate the coagulation process via the intrinsic pathway. When a contact activator, such as kaolin, is added, the test is referred to as activated PTT (aPTT). A control sample must be run with the test sample. In general, aPTT ranges from 25 to 35 seconds, and results in excess of 35 seconds are considered abnormal or prolonged. The aPTT is prolonged in cases of mild to severe deficiency of factor VIII or IX. The test result is abnormal when a given factor is 15% to 30% below its normal value.
Prothrombin Time
The prothrombin time (PT) is used to check the extrinsic pathway (factor VII) and the common pathway (factors V and X, prothrombin, and fibrinogen). For this test, tissue thromboplastin is added to the test sample to serve as the activating agent. Again, a control must be run, and results vary from one laboratory to another. In general, the normal range is 11 to 15 seconds. PT is prolonged when the plasma level of any factor is below 10% of its normal value. When the test is used to evaluate the level of anticoagulation with coumarin-like drugs the INR format is recommended. INR, a method that standardizes PT assays, is defined later in this chapter.
Platelet Count
Platelet count is used to screen for possible bleeding problems due to thrombocytopenia. Normal platelet count is 140,000 to 400,000/µL of blood. Patients with a platelet count of between 50,000 and 100,000/µL manifest excessive bleeding only with severe trauma. Patients with counts below 50,000/µL demonstrate skin and mucosal purpura and bleed excessively with minor trauma. Patients with platelet counts below 20,000/µL may experience spontaneous bleeding.
Thrombin Time
In this test, thrombin is added to the patient’s blood sample as the activating agent. It converts fibrinogen in the blood to insoluble fibrin, which makes up the essential portion of a blood clot. Again, a control must be run, and results vary from laboratory to laboratory. This test bypasses the intrinsic, extrinsic, and most of the common pathway. For example, patients with hemophilia A or fa/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


