Chapter 23
Disorders of White Blood Cells
Disorders of white blood cells (WBCs) in the dental patient can greatly influence clinical decision making as well as the specifics of care, because WBCs provide the primary defense against microbial infections and are critical for mounting an immune response (< ?xml:namespace prefix = "mbp" />

Box 23-1 Classification and Features of White Blood Cell (WBC) Dyscrasias
Leukocytosis—increased number of circulating WBCs

Three groups of WBCs are found in the peripheral circulation: granulocytes, lymphocytes, and monocytes. Of the granulocyte population, 90% is composed of neutrophils; the remainder consists of eosinophils and basophils. Circulating lymphocytes are of three types: T lymphocytes (thymus mediated), B lymphocytes (bursa-derived), and natural killer (NK) cells. Lymphocytes are subdivided by the surface markers they exhibit and by the cytokines they produce.
The primary function of neutrophils is to defend the body against certain infectious agents (primarily bacteria) through phagocytosis and enzymatic destruction. Eosinophils and basophils are involved in inflammatory allergic reactions and mediate these reactions through release of their cytoplasmic granules. Eosinophils also combat infection by parasites. T lymphocytes (T cells) are involved with the delayed, or cellular, immune reaction, whereas B lymphocytes (B cells) play an important role in the immediate, or humoral, immune system involving the production of plasma cells and immunoglobulins (IgA, IgD, IgE, IgG, and IgM). Monocytes have diverse functions that include phagocytosis, intracellular killing (especially of mycobacteria, fungi, and protozoa), and mediating of the immune and inflammatory response through the production of more than 100 substances, such as cytokines and growth factors, that increase the activity of lymphocytes. In addition, monocytes serve as antigen-presenting cells and migrate into tissues. In tissue, these antigen-presenting cells are known as dendritic cells (in lymph nodes) or Langerhans cells (in skin and mucosa). Monocytes in tissue that phagocytose microbes are known as macrophages.
Most WBCs are produced primarily in the bone marrow (granulocytes and monocytes), and these cells form several “pools” in the marrow: (1) the mitotic pool, which consists of immature precursor cells; (2) a maturing pool, which consists of cells undergoing maturation; and (3) a storage pool of functional cells, which can be released as needed.
WBCs released by the bone marrow that circulate in the peripheral blood account for only 5% of the total WBC mass and form two pools of cells: a marginal one and a circulating one. Cells in the marginal pool adhere to vessel walls and are readily available. When infection threatens the body, the storage and marginal pools can be called on to help fight the invading organisms.
Growth-promoting substances called colony-stimulating factors (CSFs) are responsible for the growth of committed granulocyte-monocyte stem cells. The major function of CSFs is to amplify leukopoiesis rather than recruit new stem cells into the granulocyte-monocyte differentiation pathway. Thus, through the local release of CSFs, the bone marrow can increase the production of granulocytes and monocytes. This process occurs in response to infection.
Lymphocytes localize primarily in three regions: lymph nodes, the spleen, and the mucosa-associated lymphoid tissue (MALT) lining the respiratory and gastrointestinal tracts. At these sites, microbial antigens are trapped and presented to B or T lymphocytes (cells). Antigens bind B cells through cell surface immunoglobulins, whereupon B cells are activated, proliferate, and produce large amounts of immunoglobulin to aid in opsonization. Antigens are presented to CD4+ (helper) T cells by major histocompatibility complex (MHC) class I molecules, and to CD8+ T cells by MHC class II molecules. CD4+ T cells activate B cells and macrophages by producing cytokines and through direct contact. CD8+ T cells kill virus-infected cells.
Leukocytosis and Leukopenia
The number of circulating WBCs normally ranges from 4400 to 11,000/µL in adults.
Many causes of leukocytosis are known. Exercise, pregnancy, and emotional stress can lead to increased numbers of WBCs in the peripheral circulation. Leukocytosis resulting from these causes is called physiologic leukocytosis. Pathologic leukocytosis can be caused by infection, neoplasia, or necrosis. Pyogenic infections induce a type of leukocytosis that is characterized by an increased number of neutrophils. If excessive numbers of immature neutrophils (stab cells) are released into the circulation in response to a bacterial infection, a shift to the left is said to have occurred. Tuberculosis, syphilis, and viral infections produce a type of leukocytosis that is characterized by increased numbers of lymphocytes. Protozoal infections often produce a type of leukocytosis that increases the numbers of monocytes. Allergies and parasitic infections caused by certain helminths increase the numbers of circulating eosinophils. Cellular necrosis increases the numbers of circulating neutrophils. Leukemia (cancer of the WBCs) is characterized by a great increase in the numbers of circulating immature leukocytes. Carcinoma of glandular tissues may cause an increase in the number of circulating neutrophils. Acute bleeding also can result in leukocytosis.
Many causes of deficient numbers of leukocytes (less than 4400/µL) in the blood are evident. Leukopenia may occur in the early phase of leukemia and lymphoma as a result of bone marrow replacement through excessive proliferation of WBCs. Leukopenia also occurs during agranulocytosis (reduction of granulocytes) and pancytopenia (decreased WBCs and RBCs) that result from toxic effects of drugs and chemicals. Leukopenia is a common complication that results from the use of chemotherapeutic (anticancer) drugs.
Cyclic Neutropenia
An important form of leukopenia involving the cyclic depression of circulating neutrophils is a disorder called cyclic neutropenia. It is associated with mutations located near the junction of exons 4 and 5 of the neutrophil elastase gene (ELA2).
Patients with leukocytosis or leukopenia may have bone marrow abnormalities that can cause thrombocytopenia. Examination of the patient’s bone marrow aspirate is important for making the final diagnosis. Infectious diseases that can cause leukocytosis and leukopenia are discussed in
Leukemia and Lymphoma
The remainder of this chapter focuses on leukemia and malignancies of lymphoid cells (lymphoma and multiple myeloma). Leukemia and lymphoma account for about 8% of all new malignancies each year in the United States, which amounts to approximately 117,080 cases per year.
Leukemia
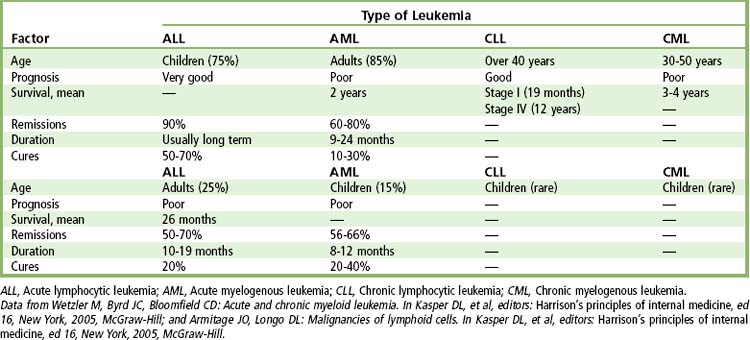
Leukemia is cancer of the WBCs that affects the bone marrow and circulating blood. It involves exponential proliferation of a clonal myeloid or lymphoid cell and occurs in both acute and chronic forms. Acute leukemia is a rapidly progressive disease that results from accumulation of immature, functionless WBCs in the marrow and blood. Chronic leukemias have a slower onset, which allows production of larger numbers of more mature (terminally differentiated), functional cells. This section focuses on four types of leukemia: (1) acute lymphocytic leukemia (ALL), (2) acute myelogenous leukemia (AML), (3) chronic lymphocytic leukemia (CLL), and (4) chronic myelogenous leukemia (CML).
Leukemia occurs in all races, at any age, at an incidence of 12.3 per 100,000.
Leukemia is much more common in adults than in children, with more than half of all cases occurring after age 65 years. The most common types of leukemia in adults are acute myelogenous leukemia, with an estimated 12,330 new cases in 2010, and chronic lymphocytic leukemia, with some 14,990 new cases in 2010.
The cause of leukemia remains unknown. Increased risk is associated with large doses of ionizing radiation, certain chemicals (benzene), and infection with specific viruses (e.g., Epstein-Barr virus [EBV], human lymphotropic virus [HTLV]-1). Cigarette smoking and exposure to electromagnetic fields also have been proposed to be causative.
Acute Myelogenous Leukemia
Definition
AML is a neoplasm of myeloid (immature) WBCs, which demonstrate uncontrolled proliferation in the bone marrow space and subsequently appear in the peripheral blood.
Epidemiology
In 2010, AMLs accounted for 28.6% of all leukemias.
Etiology
AML arises de novo in younger adults or secondarily in elderly persons as a consequence of myelodysplasia. Environmental factors such as tobacco smoke, benzene-containing products, chemotherapies for cancer, and radiation exposure appear to be risk factors.
Pathophysiology and Complications
AML has a sudden onset and leads to death in 1 to 3 months if left untreated.
Clinical Presentation
Signs and Symptoms
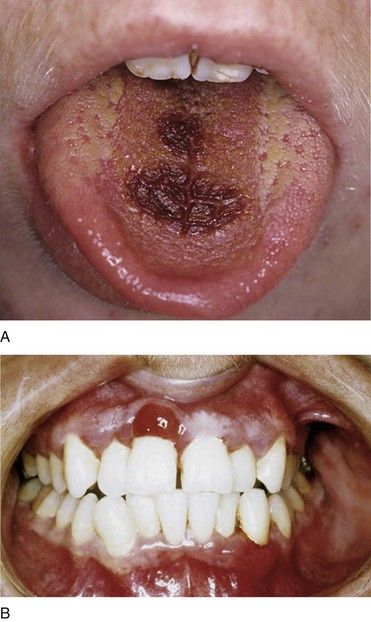
AML produces a leukemic infiltration of marrow and organs that causes cytopenia and diverse nonspecific signs and symptoms, including fatigue, easy bruising, and bone pain. Many patients complain of flulike symptoms for 4 to 6 weeks before the diagnosis. Anemia and thrombocytopenia usually manifest as malaise, pallor, dyspnea on exertion, and bleeding and small hemorrhage (petechiae, ecchymoses) in the skin and mucous membranes (
Laboratory Findings
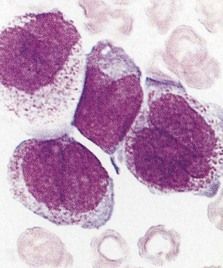
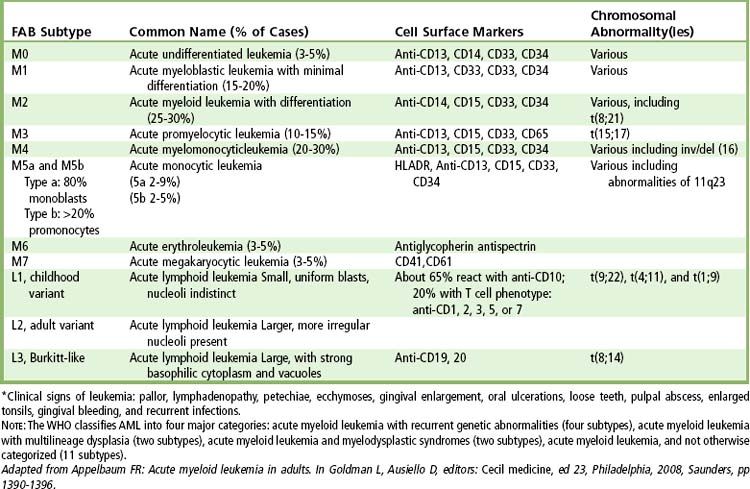
The diagnosis of leukemia is made through examination of peripheral blood and bone marrow stained with Wright-Giemsa. Cytochemical staining, immunophenotyping, and cytogenetic analyses are used to characterize the type and subtype, to allow for specific treatment approaches, and to detect residual disease after therapy is provided. Granulocytopenia and thrombocytopenia are common.
The diagnosis of AML is made when myeloblasts are found in the bone marrow or peripheral blood at a rate of at least 20%. Myeloblasts stain positive for myeloperoxidase and are immunotype-positive for several of the following markers: CD13, CD33, CD34, CD65, and CD117.
Acute Lymphoid Leukemia
Definition
ALL is the result of uncontrolled monoclonal proliferation of immature lymphoid cells in the bone marrow and peripheral blood. These neoplastic cells may also expand in the lymph nodes, liver, spleen, or CNS.
Epidemiology
In 2010 there were 5330 cases of ALL reported in the United States.
Etiology
Although environmental, infectious, and genetic factors are considered likely causes of the disease, causal links for ALL have not been established. The disease is 18- to 20-fold more common in patients with Down syndrome (trisomy 21). Cytogenetic studies frequently display the Philadelphia chromosome [t(9;22)], a shortened chromosome 22, as a result of translocation of genes between the long arms of chromosomes 9 and 22. About 5% of children and 25% of adults with ALL have cytogenetics showing the Philadelphia chromosome. Patients with the Philadelphia chromosome have slightly lower complete remission rates and greatly reduced remission durations. Other chromosomal anomalies are also common.
Pathophysiology and Complications
Similar to AML, ALL results in suppression of normal hematopoiesis, leaving patients susceptible to excessive bleeding, anemia, poor healing, and infection after surgical procedures have been performed.
Clinical Presentation
Signs and Symptoms
The clinical presentation of ALL can be acute or insidious. Presenting signs and symptoms relate to anemia, thrombocytopenia, fever, and neutropenia. Frequently, bone and joint pain have effects on walking. In one large study one third of the patients presented with infection or fever and one third with hemorrhagic episodes, and over half of the patients with enlargement of the liver, spleen, and lymph nodes.
Laboratory Findings
ALL is diagnosed when massive replacement of the bone marrow space with leukemic blast cells is observed.
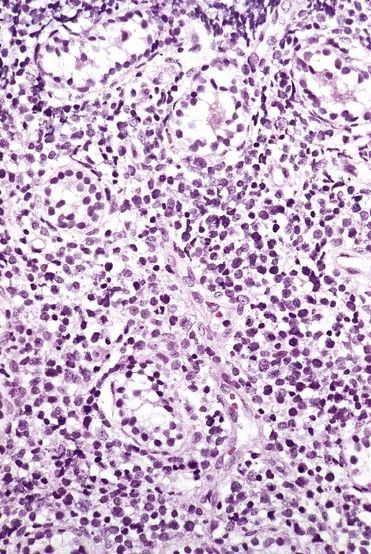
FIGURE 23-3 Peripheral blood smear of acute lymphoblastic leukemia.
(From Hoffbrand AV, Pettit JE: Color atlas of clinical hematology, ed 4, London, 2010, Mosby.)
According to the French-American-British Cooperative Group, three distinct subtypes are based on type and size of neoplastic lymphocytes: L1 (cells small and homogeneous), L2 (cells pleomorphic and often large), and L3 (cells homogeneous and of medium size with dispersed chromatin).
Medical Management of Acute Leukemia
The ability to cure a patient of acute leukemia is related to tumor burden and the rapid elimination of malignant WBCs. Normal bone marrow consists of 0.3% to 5% blast cells. Patients with acute leukemia have 100-fold more (about a trillion) blast cells. Once effective chemotherapy has been given, the number of blast cells is reduced from trillions to billions, leukemic cells can no longer be detected, and the patient is said to be in remission. With a 5-day generation time for the remaining undetectable leukemic cell mass, 10 doublings in 50 days could restore the leukemic cell mass to a trillion cells, and the patient would again show signs and symptoms of leukemia. This would constitute a short remission with relapse.
Chemotherapy for acute leukemia consists of three phases. The purpose of the first phase (induction) is to hit hard and induce a state of remission by killing tumor cells with cytotoxic agents. Agents used to treat the acute leukemias are shown in
TABLE 23-2 Classes of Drugs Used to Treat Leukemia
| Drug Class | Chemotherapeutic Agents | Mechanism of Action |
|---|---|---|
| Alkylating agents | Busulfan, carmustine, cyclophosphamide, dacarbazine, lomustine nitrogen mustard Derivative: chlorambucil | Produce alkyl radicals, causing cross-linking of DNA and inhibition of DNA synthesis in rapidly replicating tumor cells |
| Antibiotics | Bleomycin, daunorubicin, doxorubicin, idarubicin, mitomycin C | Disrupt cellular functions, such as RNA synthesis, or inhibit mitosis |
| Antimetabolites | Folic acid analogues: methotrexate | Disrupt enzymatic processes or nucleic acid synthesis |
| Purine analogues: cladribine, fludarabine, fluorouracil 6-mercaptopurine, thioguanine | ||
| Pyrimidine nucleoside analogues: arabinosyl cytosine (Ara-C, cytarabine) | ||
| Biologicals | Interferon alfa | Causes a direct antiproliferative effect on CML progenitor cells |
| Rituximab Alemtuzumab |
Monoclonal antibody to CD20 Monoclonal antibody to CD52 |
|
| All-trans retinoic acid (ATRA) [tretinoin] | Binds antigen target on malignant lymphocyte | |
| Induces differentiation and apoptosis of malignant promyelocytes in APML | ||
| Enzymes | Asparaginase | Inhibits synthesis of asparagines, which is required for protein synthesis in leukemic lymphoblasts |
| Mitotic inhibitors | Vincristine, vinblastine | Act as mitotic spindle inhibitors causing metaphase arrest |
| Etoposide | Topoisomerase II inhibitor | |
| Steroid | Prednisone | Hormone that has antiinflammatory and antilymphocytic properties |
| Newer agents | ||
| Agents in clinical trials |
APML, Acute promyelocytic leukemia; CML, Chronic myelogenous leukemia.
Patients are cured of leukemia when no leukemic cells remain. Long-term survival occurs when the leukemic cell mass is greatly reduced and is kept from increasing over a long period. In general, once a patient relapses, a second remission is more difficult to induce, and if it occurs, it will be of a shorter duration. Bone marrow transplantation (BMT) generally is reserved for patients younger than 45 years of age and for children and young adults who relapse when a suitable sibling match is available (allogeneic).
Treatment of patients with AML is shown in
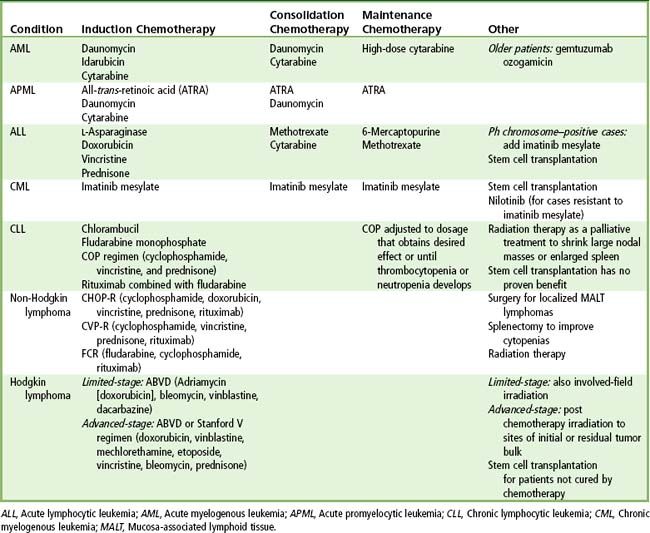
Treatment for ALL is shown in
Another concern related to treatment of patients with acute leukemia is that leukemic cells can migrate to areas in the body where chemotherapeutic agents cannot reach them. These areas are called sanctuaries, and they require special treatment. The most important sanctuary in patients with ALL is the CNS. Thus, patients with ALL are treated with systemic chemotherapy plus high-dose methotrexate intravenously and cytarabine or intrathecal methotrexate and radiation to the cranium plus high-dose systemic chemotherapy. Another important sanctuary (in males) is the testes.
Oral Manifestations of Acute Leukemia
Leukemic patients are prone to develop gingival enlargement, ulceration, and oral infection. Localized or generalized gingival enlargement is caused by inflammation and infiltration of atypical and immature WBCs (see
A localized mass of leukemic cells (in the gingiva or other sites) is specifically known as a granulocytic sarcoma or chloroma. These extramedullary tumors have been observed in the maxilla and the palate.
Chronic Myelogenous Leukemia
Epidemiology
CML has an incidence of 1 to 1.5 cases per 100,000 population, with 4870 cases reported for 2010 in the United States.
Etiology
The etiology is unknown, but radiation exposure increases risk for the disease. The genetic defect consists of translocation of the cellular oncogene ABL (Abelson leukemia virus gene) from chromosome 9 to the BCR (breakpoint cluster region) gene of chromosome 22 and a reciprocal translocation of part of BCR from chromosome 22 to the ABL gene in chromosome 9. A shortened chromosome 22, the Philadelphia (Ph) chromosome, results from the translocations and is evident in more than 90% of cases of CML.
Pathophysiology and Complications
CML progresses slowly through a chronic phase for 3 to 5 years and then moves on to an accelerated phase, followed by a blast phase (or crisis). More than 90% of the patients when first diagnosed are in the chronic phase of the disease. During the chronic phase of CML, leukemic cells are functional; thus, infection is not a major problem. However, once transformation to the blastic stage has occurred, the leukemic cells are immature and nonfunctional. As a result, anemia, thrombocytopenia, and infection become problems. In about 25% of patients with CML per year exhibit progression to the blast phase of the disease 6 to 12 months after diagnosis. The blast phase is characterized by 30% or more leukemic blast cells in the peripheral blood or marrow.
Clinical Presentation
Signs and Symptoms
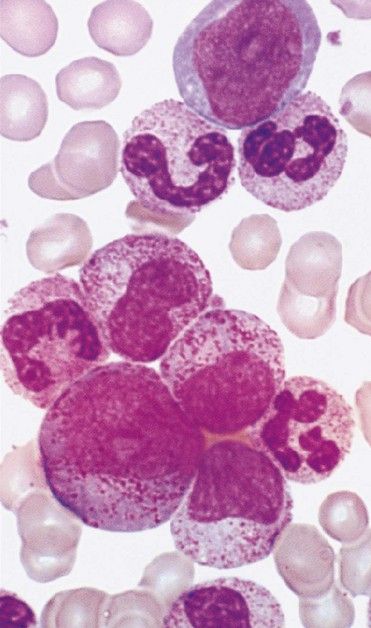
In nearly 90% of patients, CML is diagnosed during the chronic phase. Up to half of these patients are asymptomatic, and diagnosis is based on their complete blood cell count. Common symptoms are fatigue, weakness, abdominal (upper left quadrant) pain, abdominal fullness, weight loss, night sweats due to anemia, an enlarged and painful spleen (splenomegaly), and altered hematopoiesis. Hyperviscosity of the blood may cause a stroke.
Laboratory Findings
Patients are identified by marked elevation of their WBC count during routine examination (
Medical Management
Patients with CML were historically treated during the chronic phase with hydroxyurea or busulfan; this approach resulted in good symptom and blood count control, along with significant toxicity. Interferon-α or imatinib mesylate (Gleevec), an inhibitor of tyrosine kinase, is widely used today.
Chronic Lymphocytic Leukemia
Epidemiology
CLL is the most common type of leukemia in adults. In 2010 there were 14,990 cases of CLL reported in the United States. The incidence rate is 4 to 5.3 cases per 100,000.
Etiology
The etiology of CLL is unknown, and risk factors are more related to familial inheritance than to exposure to harmful environmental agents. Neoplastic B cells have various genetic aberrations, most commonly gene deletions (e.g., on chromosome 11, 12, or 17) that lead to loss of cell cycle control.
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


