CHAPTER 22 Histamine and Histamine Antagonists
HISTAMINE
Histamine was the first autacoid to be discovered. After its synthesis in 1907, a series of studies by Dale and Laidlaw10 of the pharmacologic properties of histamine suggested that this substance might be involved in inflammatory and anaphylactic reactions. Dale and Laidlaw10 observed that the local application of histamine caused redness, swelling, and edema, mimicking a mild inflammatory reaction. They also determined that large doses of histamine given systemically produced profound vascular changes similar to those seen in shock of traumatic or anaphylactic origin. Although the presence of histamine in animal tissues had been suggested, it was not until 1927 that histamine was conclusively shown to be a natural constituent of mammalian tissues and not the result of bacterial action.2 This finding provided important support for the work of Lewis and Grant,23 who had shown earlier that a histamine-like substance (“H substance”) was released in the skin after various injuries, including antigen-antibody reactions.
These early studies and the studies that followed established that histamine is involved in various pathophysiologic phenomena seen after injury to tissue. Since then, a large amount of detailed information regarding the synthesis, storage, release, and actions of histamine has been generated. Despite this knowledge base, our understanding of the role of histamine in the complex response of cells to injurious stimuli and the relation of this compound to other autacoids is meager. It is increasingly evident that this ubiquitous amine is involved in physiologic processes other than reaction to injury. These processes include gastric secretion,8 neurotransmission in the central nervous system (CNS),36,43 and local control of the microcirculation.55
Formation, Distribution, and Release
Histamine is widely distributed in nature and is found in plants, bacteria, and animals. Nearly all mammalian tissues contain histamine or have the ability to form it. The histamine content of different tissues varies greatly. In humans and most other mammals, the highest concentrations are found in lung, skin, and intestinal mucosa; organs such as the pancreas, spleen, liver, and kidney have a low histamine content (Table 22-1). The physiologic significance of this pattern of distribution is unknown. Although some tissue histamine may be derived from dietary sources or synthesized by bacteria in the gastrointestinal tract, most of it seems to be formed in situ.
TABLE 22-1 Distribution and Content of Histamine in Various Human Tissues and Cells
| TISSUE OR CELL | HISTAMINE CONTENT* |
|---|---|
| Lung | 33 ± 10 |
| Mucosa (nasal) | 15.6 (range 5-38.5) |
| Stomach | 14 ± 4 |
| Duodenum | 14 ± 0.9 |
| Skin (face) | 30.4 |
| Skin (abdomen) | 6.6 |
| Pancreas | 4.8 ± 1.5 |
| Spleen | 3.4 ± 1 |
| Bone marrow | 3.3 ± 1.5 |
| Kidney | 2.5 ± 1.2 |
| Liver | 2.2 ± 0.8 |
| Heart | 1.6 ± 0.1 |
| Thyroid | 1 ± 0.1 |
| Skeletal muscle | 0.9 ± 0.1 |
| Peripheral nerves | 2-11 |
| CNS tissue | 0-0.2 |
| Whole blood | 16-89 µg/L |
| Plasma | 2.6 µg/L (range 0-15) |
| Basophils | 1080 µg/109 cells |
| Eosinophils | 160 µg/109 cells |
| Neutrophils | 3.0 µg/109 cells |
| Lymphocytes | 0.6 µg/109 cells |
| Platelets | 0.009 µg/109 platelets |
CNS, Central nervous system.
* Means or means ± standard error expressed as µg/g unless otherwise indicated.
From Van Arsdel PP Jr, Beall GN: The metabolism and functions of histamine, Arch Intern Med 106:714-733, 1960. Copyright 1960, American Medical Association.
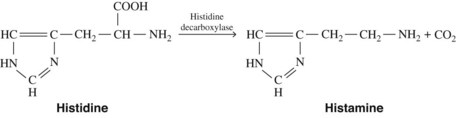
Histamine is synthesized in mammalian tissues by the intracellular decarboxylation of the amino acid histidine (Figure 22-1). This conversion may be catalyzed either by aromatic l-amino acid decarboxylase or by histidine decarboxylase. Histidine decarboxylase is specific for l-histidine, requires pyridoxal phosphate, and seems to be primarily responsible for the synthesis of histamine in humans.
Histamine is found in most tissues in the mast cell and in blood in a related cell, the basophil.39,44 These cells synthesize histamine and store it as a proteinaceous complex with heparin or chondroitin sulfate in membrane-bound secretory granules. Histamine in this form is physiologically inactive but can be discharged from the cell by a process called exocytosis, or degranulation. The first step in this process is activation of the cell by an appropriate stimulus. When the cell is activated, a complex series of events leading to degranulation occurs. These events require an increase in cytosolic Ca++ and metabolic energy and involve activation of Ca++-dependent protein kinases (protein kinase C and Ca++/calmodulin-dependent protein kinase), assembly of microtubules, and, finally, fusion of the perigranular membrane with the cell membrane. The granule contents are released into the extracellular environment and dissociate to yield histamine, heparin, several proteases, and chemoattractants such as tumor necrosis factor α. In addition to release of these preformed mediators, activation of mast cells activates phospholipase A in the cell membrane, which hydrolyzes phospholipid esters to yield arachidonic acid. The arachidonic acid is metabolized by the cyclooxygenase pathway to various prostaglandins and thromboxanes and by the lipoxygenase pathway to various leukotrienes.
A small amount of histamine not stored in mast cells or basophils is found in several sites. One of these sites is certain neurons in the hypothalamus. The function of these histaminergic neurons is unknown. Another site of non–mast cell histamine is the enterochromaffin-like cell in the gastric mucosa. These cells synthesize and release histamine, which subsequently stimulates gastric acid secretion by mucosal parietal cells (see Chapter 33). Certain neoplasms collectively known as carcinoid can also secrete various bioactive substances, including histamine.9 These substances likely contribute to the so-called carcinoid syndrome, a prominent feature of which is cutaneous flushing and bronchoconstrictive attacks similar to that seen after the intravascular administration of histamine.
Tissue injury
Any physical or chemical agent that nonspecifically injures tissue, particularly skin or mucosa, causes the immediate release of histamine from mast cells in the affected area. Depending on the severity of injury, histamine continues to be released for several minutes and seems to be largely responsible for the initial sharp increase in vascular permeability that is characteristic of acute inflammation. This histamine-dependent change in permeability is transient (≤30 minutes) but is followed in 2 to 4 hours by a more prolonged increase in permeability lasting up to 4 hours. Although inhibitors of histamine release or inhibitors of the subsequent action of histamine can block the initial phase of vascular permeability after injury, they have little effect on the secondary or delayed phase, suggesting that autacoids or factors other than histamine mediate the secondary phase.54 The mechanism by which a nonspecific injury triggers mast cell degranulation is unclear. It could depend on direct physical damage to mast cells, or alternatively it may involve the initial production of factors such as activated complement components or vasoactive polypeptides, which stimulate histamine release.21,31
Allergic reactions
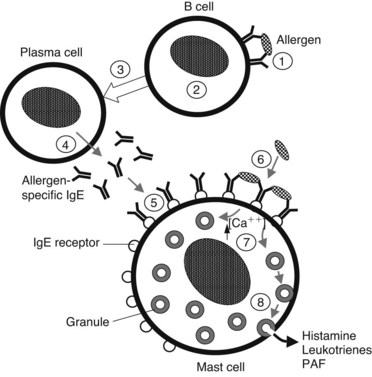
Presentation of a specific antigen to a previously sensitized subject can trigger immediate allergic reactions, ranging in intensity from mild (localized edema, erythema, and itching) to severe (marked decrease in blood pressure and bronchospasm). The pathophysiologic manifestations of such reactions are caused largely by the release of histamine (Figure 22-2). This release occurs as a consequence of the binding of specific antigens to allergen-specific reaginic (IgE) antibodies attached to the plasma membranes of mast cells and basophils transmembrane high-affinity receptors termed FcεRI.29 Binding of antigen and antibody may cause conformational changes in the membrane, leading to an increase in Ca++ permeability. In any case, this antigen-antibody interaction is an appropriate stimulus for the series of events leading to degranulation of these cells.
Drugs and other foreign compounds
Although drugs that are antigenic (e.g., penicillin) can cause histamine release, a large group of drugs and other chemicals can trigger histamine release directly without a requirement for previous sensitization through an immune response. For convenience, these agents can be classified as basic histamine releasers, macromolecular compounds, and enzymes.21 The basic histamine releasers include aliphatic and arylalkyl amines, amides, amidines, diamidines, quaternary ammonium compounds, alkaloids, piperidine derivatives, pyridinium compounds, antimalarial drugs, dyes, and basic polypeptides. The prototypic histamine releaser compound 48/80 and numerous therapeutic agents of interest, such as tubocurarine and morphine, fall into this category.
Metabolism
Histamine of either exogenous or endogenous origin is rapidly inactivated by two routes.41 The more important of these is methylation of the imidazole ring by the enzyme histamine-N-methyltransferase, which is widely distributed throughout the body. The resultant product is converted to methylimidazole acetic acid by monoamine oxidase. The other route involves the oxidative deamination of histamine by diamine oxidase to produce imidazole acetic acid, much of which is subsequently conjugated with ribose. All these metabolites are inactive and, along with a small amount of free histamine, are excreted by the kidney.
Pharmacologic Effects
The existence of compounds that can selectively block the actions of histamine strongly supports the existence of four histamine receptors, H1, H2, H3, and H4.16,18 Although the first two seem to be unique (i.e., have selective agonists and antagonists), the H3 and H4 receptors share a degree of homology and are more difficult to distinguish pharmacologically from one another.47 All four histamine receptors are G protein–linked receptors. H1 receptors are associated with Gq/11, and stimulation of these receptors leads to an increase in intracellular Ca++ and Ca++-dependent protein kinase activity. H2 receptors are linked to Gs, and stimulation of these receptors brings about an increase in intracellular cyclic adenosine 3′,5′-monophosphate (cAMP). H3 and H4 receptors seem to stimulate Gi/o proteins, leading to reduced cAMP (see Chapters 1 and 5 for more details on the specifics of signal transduction involving G proteins).
H1 receptors primarily mediate effects on smooth muscle, leading to vasodilation, increased vascular permeability, and contraction of nonvascular smooth muscle. These effects are blocked by the “classic” antihistamines, such as pyrilamine. H2 receptors mediate the stimulation of gastric acid secretion. H2 receptors may be involved in other effects, such as direct cardiac stimulation and vasodilation seen after high doses of histamine, based on studies showing that H2-blocking agents specifically antagonize these effects.5,6
H3 receptors have been identified in various cells, including presynaptic sites on histaminergic neurons in the CNS and prejunctional sites in gastric mucosa, cardiovascular system, and the enterochromaffin-like cells of the stomach. The function of H3 receptors is not firmly established, but these receptors seem to antagonize stimulatory effects mediated by the activation of H1 receptors. Their prejunctional location suggests a role as autoregulatory receptors in many tissues. Activation of H3 receptors on adrenergic nerve terminals in the myocardium inhibits the release of norepinephrine.19,22 The more recently identified H4 histamine receptor is found primarily on cells of hematopoietic origin, particularly mast cells, basophils, and eosinophils. Activation of H4 receptors on these cells induces changes in cell shape, chemotaxis, and upregulation of various cell-adhesion molecules.17,24
Cardiovascular system
The effect of histamine on the terminal vasculature can be illustrated by injection of 10 to 20 µg of the amine into the skin. At the site of the injection, there is first immediate reddening, reflecting vasodilation. This reddening is followed shortly by a zone of erythema, or “flare,” extending as an irregular halo for 1 cm or more beyond the original red spot. The flare is presumed to be caused by reflex vasodilation of adjacent small vessels, resulting from the axon reflex, and is abolished by disruption of the peripheral sensory nerves. Finally, the central spot is replaced by a disk of localized edema, or wheal, resulting from increased capillary permeability, and is accompanied by pain and itching. These events constitute the classic triple response first described by Lewis and Grant.23 A similar response is elicited by the intradermal injection of antigen in a sensitized individual.
In an intact animal, histamine can cause cardiac stimulation, principally the result of reflex mechanisms triggered by the histamine-induced decrease in peripheral vascular resistance. Histamine also has some direct positive chronotropic and inotropic effects on the heart. The receptors responsible are largely H2 receptors.22
Exocrine glands
The relationships among histamine and the potent gastric secretagogues acetylcholine and the polypeptide hormone gastrin are complex (see Figure 33-1). As previously noted, histamine is synthesized by the enterochromaffin-like cells in the gastric mucosa, and its release from these cells is triggered by either gastrin or acetylcholine. The fact that H2 receptor antagonists inhibit stimulation of gastric secretion by histamine, gastrin, or acetylcholine lends support to the possibility that the latter two agents act by releasing histamine from enterochromaffin-like cells, which acts directly on the parietal cells through H2 receptors.33 Identification of specific receptors for gastrin and acetylcholine on the parietal cell, plus an ability of gastrin to augment (although moderately) histamine-induced secretion, indicates, however, that gastrin can also cause release of H+ by acting directly on parietal cells and independently of histamine.
HISTAMINE ANTAGONISTS
Early interest in histamine as a mediator of certain pathologic processes of an allergic nature spurred interest in agents that could block histamine. The first such compound, a derivative of phenoxyethylamine, was reported by Bovet and Staub in 1937.5 Although this substance could adequately protect guinea pigs against injected histamine or anaphylactic shock, it was too toxic for human use. Other, less toxic compounds with antihistaminic activity were immediately sought. By 1946, numerous compounds with therapeutically useful properties had been found, including phenbenzamine, pyrilamine, diphenhydramine, and tripelennamine. During the following 20 years, hundreds of other compounds with antihistaminic properties were developed.
As the pharmacologic properties of antihistamines were studied, it became apparent that they had no effect on histamine-induced gastric acid secretion. In 1972, a potent antagonist of histamine-induced gastric secretion, burimamide, was discovered.3 The subsequently developed congeners of burimamide—the H2 receptor antagonists—quickly became an important new class of therapeutic agents and tools for further investigation of the role of histamine in health and disease.
Antagonists of H3 and H4 receptors have more recently been identified. In animal stu/>
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


