Bone Loss and Patterns of Bone Destruction
The height and density of the alveolar bone are normally maintained by an equilibrium, regulated by local and systemic influences, between bone formation and bone resorption.12,14 When resorption exceeds formation, both bone height and bone density may be reduced (see Video 21-1: Vertical Bone Loss) .
.
Bone Destruction Caused by the Extension of Gingival Inflammation
The transition from gingivitis to periodontitis is associated with changes in the composition of bacterial plaque. In advanced stages of disease, the number of motile organisms and spirochetes increases, whereas the number of coccoid rods and straight rods decreases.34 The cellular composition of the infiltrated connective tissue also changes with increasing severity of the lesion (see Chapter 7). Fibroblasts and lymphocytes predominate in stage 1 gingivitis, whereas the number of plasma cells and blast cells increases gradually as the disease progresses. Seymour and colleagues58,59 have postulated a stage of “contained” gingivitis in which T lymphocytes are preponderant; they think that, as the lesion becomes a B-lymphocyte lesion, it becomes progressively destructive.
Heijl and coworkers22 were able to convert, in experimental animals, a confined, naturally occurring chronic gingivitis into a progressive periodontitis by placing a silk ligature into the sulcus and tying it around the neck of the tooth. This induced ulceration of the sulcular epithelium, a shift in the inflammatory infiltrate from predominantly plasma cells to predominantly polymorphonuclear leukocytes, and osteoclastic resorption of the alveolar crest. The recurrence of episodes of acute destruction over time may be one mechanism that leads to progressive bone loss in individuals with marginal periodontitis.
The extension of inflammation to the supporting structures of a tooth may be modified by the pathogenic potential of plaque and the resistance of the host. The latter includes immunologic activity and other tissue-related mechanisms, such as the degree of fibrosis of the gingiva, probably the width of the attached gingiva, and the reactive fibrogenesis and osteogenesis that occur peripheral to the inflammatory lesion.52
Histopathology.
Gingival inflammation extends along the collagen fiber bundles and follows the course of the blood vessels through the loosely arranged tissues around them into the alveolar bone67 (Figure 21-1). Although the inflammatory infiltrate is concentrated in the marginal periodontium, the reaction is a much more diffuse one, often reaching the bone and eliciting a response before evidence of crestal resorption or loss of attachment exists.41 In the upper molar region, inflammation can extend to the maxillary sinus, thereby resulting in thickening of the sinus mucosa.40
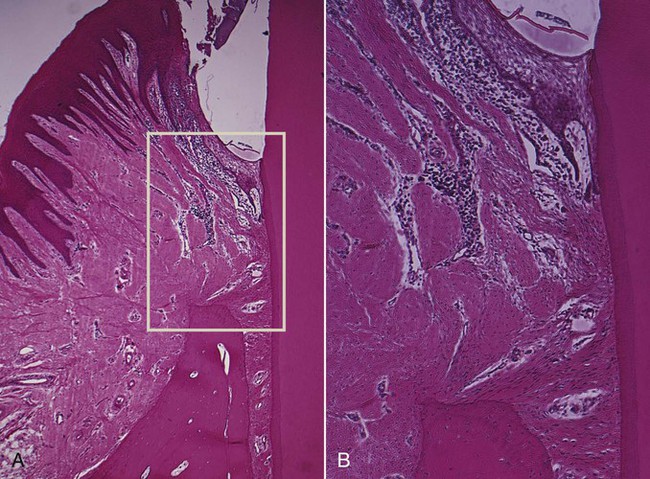
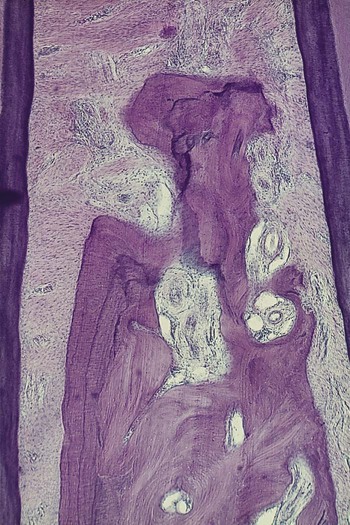
Interproximally, inflammation spreads to the loose connective tissue around the blood vessels, through the fibers, and then into the bone through vessel channels that perforate the crest of the interdental septum at the center of the crest (Figure 21-2), toward the side of the crest (Figure 21-3), or at the angle of the septum. In addition, inflammation may enter the bone through more than one channel. Less frequently, the inflammation spreads from the gingiva directly into the periodontal ligament and from there into the interdental septum1 (Figure 21-4).
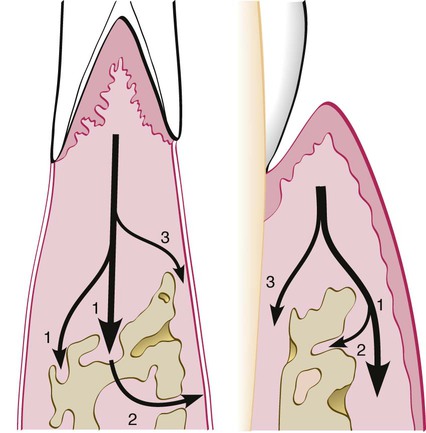
Facially and lingually, inflammation from the gingiva spreads along the outer periosteal surface of the bone (see Figure 21-4) and penetrates into the marrow spaces through vessel channels in the outer cortex.
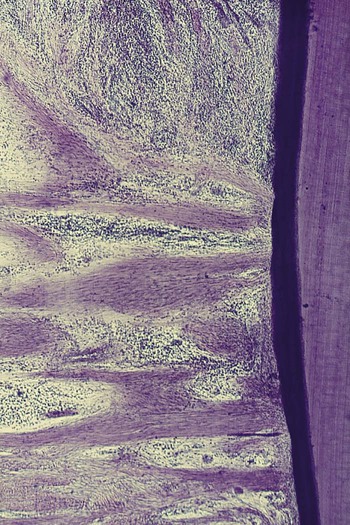
Along its course from the gingiva to the bone, the inflammation destroys the gingival and transseptal fibers, reducing them to disorganized granular fragments interspersed among the inflammatory cells and edema.45 However, there is a continuous tendency to recreate transseptal fibers across the crest of the interdental septum farther along the root as the bone destruction progresses (Figure 21-5). As a result, transseptal fibers are present, even in cases of extreme periodontal bone loss.
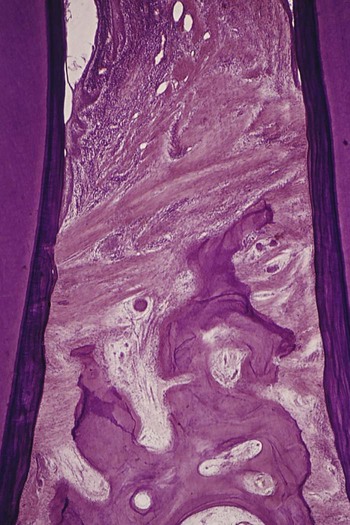
The dense transseptal fibers form a firm covering over the bone that is encountered during periodontal flap surgery after the superficial granulation tissue is removed.50
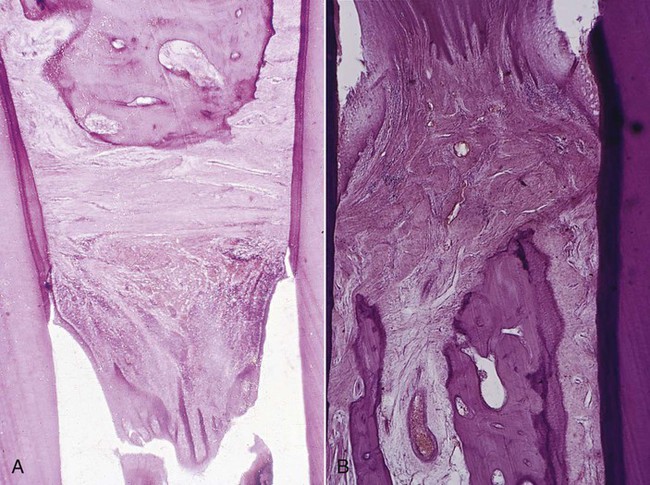
After inflammation reaches the bone via extension from the gingiva (Figure 21-6), it spreads into the marrow spaces and replaces the marrow with a leukocytic and fluid exudate, new blood vessels, and proliferating fibroblasts (Figure 21-7). Multinuclear osteoclasts and mononuclear phagocytes increase in number, and the bone surfaces appear to be lined with Howship lacunae (Figure 21-8).
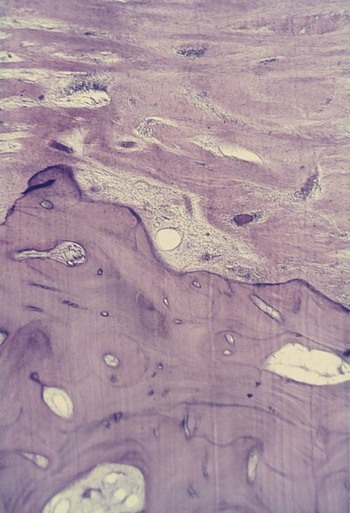
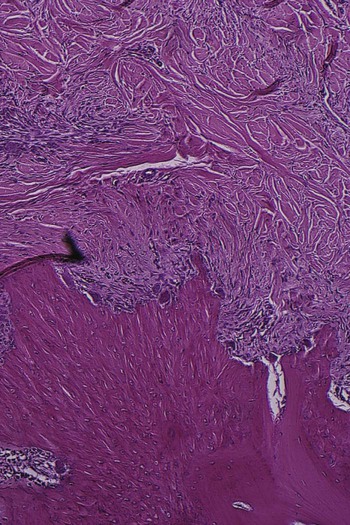
Bone destruction in periodontal disease is not a process of bone necrosis.27 It involves the activity of living cells along viable bone. When tissue necrosis and pus are present in periodontal disease, they occur in the soft-tissue walls of periodontal pockets rather than along the resorbing margin of the underlying bone.
The amount of inflammatory infiltrate correlates with the degree of bone loss but not with the number of osteoclasts. However, the distance from the apical border of the inflammatory infiltrate to the alveolar bone crest correlates with both the number of osteoclasts on the alveolar crest and the total number of osteoclasts.51 Similar findings have been reported in experimentally induced periodontitis in animals.32
Radius of Action
Garant and Cho10 suggested that locally produced bone resorption factors may need to be present in the proximity of the bone surface to exert their action. Page and Schroeder,49 on the basis of Waerhaug’s measurements made on human autopsy specimens,64,65 postulated a range of effectiveness of about 1.5 mm to 2.5 mm in which bacterial plaque can induce loss of bone. Beyond 2.5 mm, there is no effect; interproximal angular defects can appear only in spaces that are wider than 2.5 mm, because narrower spaces would be destroyed entirely. Tal61 corroborated this with measurements in human patients.
Large defects that greatly exceed a distance of 2.5 mm from the tooth surface (as described in aggressive types of periodontitis) may be caused by the presence of bacteria in the tissues.6,10,54
Rate of Bone Loss
In a study of Sri Lankan tea laborers with no oral hygiene and no dental care, Löe and colleagues36 found the rate of bone loss to average about 0.2 mm per year for facial surfaces and about 0.3 mm per year for proximal surfaces when periodontal disease was allowed to progress untreated. However, the rate of bone loss may vary, depending on the type of disease present. Löe and colleagues35 also identified the following three subgroups of patients with periodontal disease on the basis of the interproximal loss of attachment and tooth mortality (loss of attachment can be equated with loss of bone, although attachment loss precedes bone loss by about 6 to 8 months17):
1. Approximately 8% of persons had a rapid progression of periodontal disease that was characterized by a yearly loss of attachment of 0.1 mm to 1.0 mm.
2. Approximately 81% of individuals had moderately progressive periodontal disease with a yearly loss of attachment of 0.05 mm to 0.5 mm.
3. The remaining 11% of persons had minimal or no progression of destructive disease with a yearly loss of attachment of 0.05 mm to 0.09 mm.
Periods of Destruction
Periods of destructive activity are associated with subgingival ulceration and an acute inflammatory reaction that results in the rapid loss of alveolar bone49,56; it has been hypothesized that this coincides with the conversion of a predominantly T-lymphocyte lesion to one with a predominantly B-lymphocyte–plasma cell infiltrate.59 Microbiologically, these lesions are associated with an increase in the loose, unattached, motile, gram-negative, anaerobic pocket flora, whereas periods of remission coincide with the formation of a dense, unattached, nonmotile, gram-positive flora with a tendency to mineralize.43
It has also been suggested that the onset of periods of destruction coincide with tissue invasion by one or several bacterial species and that this is followed by an advanced local host defense that controls the attack.54
Mechanisms of Bone Destruction
The factors involved in bone destruction in periodontal disease are bacterial and host mediated. Bacterial plaque products induce the differentiation of bone progenitor cells into osteoclasts and stimulate gingival cells to release mediators that have the same effect.21,57 Plaque products and inflammatory mediators can also act directly on osteoblasts or their progenitors, thereby inhibiting their action and reducing their numbers.
In addition, in patients with rapidly progressing diseases (e.g., aggressive periodontitis), bacterial microcolonies or single bacterial cells have been found between collagen fibers and over the bone surface, which suggests a direct effect.6,9,57
When injected intradermally, prostaglandin E2 induces the vascular changes that are seen with inflammation; when injected over a bone surface, prostaglandin E2 induces bone resorption in the absence of inflammatory cells, with few multinucleated osteoclasts.18,26 In addition, nonsteroidal anti-inflammatory drugs (e.g., flurbiprofen, ibuprofen) inhibit prostaglandin E2 production, thereby slowing bone loss in naturally occurring periodontal disease in beagle dogs and humans. This effect occurs without changes in gingival inflammation, and it rebounds 6 months after the cessation of drug administration.25,68 (For more information about the host-mediated mechanisms of bone destruction, see Chapter 25.)
Bone Formation in Periodontal Disease
Areas of bone formation are also found immediately adjacent to sites of active bone resorption and along trabecular surfaces at a distance from the inflammation in an apparent effort to reinforce the remaining bone (i.e., buttressing bone formation). This osteogenic response is clearly found in experimentally produced periodontal bone loss in animals.7 In humans, it is less obvious, but it has been confirmed by histometric4,5 and histologic studies.13
The periods of remission and exacerbation (or inactivity and activity, respectively) appear to coincide with the quiescence or exacerbation of gingival inflammation as manifested by changes in the extent of bleeding, the amount of exudate, and the composition of bacterial plaque (see Chapter 23).
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


