CHAPTER 2 Pharmacokinetics
The Absorption, Distribution, and Fate of Drugs
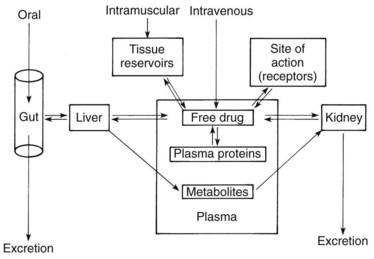
When the magnitude of a drug’s pharmacologic effect is quantified as a function of dose, the tacit assumption is that the drug concentration vicinal to the site of action is linearly related to the amount administered. Although this assumption may strictly apply to an in vitro test, it ignores the temporal factors that modify drug effects in vivo. Drug concentrations are rarely static; they increase and decrease as dictated by the processes of absorption, distribution, metabolism, and excretion. This chapter examines these processes (Figure 2-1) and how they influence the passage of drugs through the body.
PASSAGE OF DRUGS ACROSS MEMBRANES
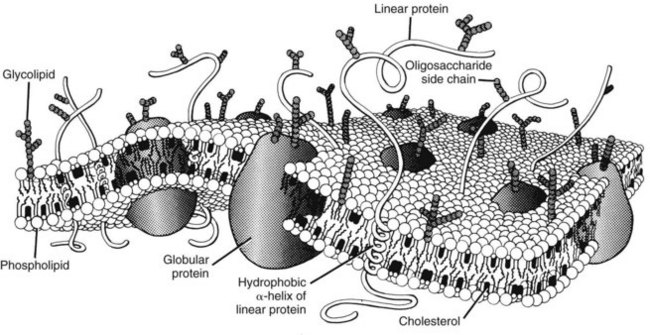
For a drug to be absorbed, reach its site of action, and eventually be eliminated, it must cross one or more biologic membrane barriers. These may consist of a single plasma membrane or constitute a layer of closely packed cells. Because such barriers to drugs behave similarly, the cell membrane can serve as a prototype for all. The plasma membrane is composed of a bimolecular sheet of lipids (primarily phospholipids and cholesterol) with proteins interspersed throughout and extending beyond the lipid phase of the membrane (Figure 2-2).8,51 The presence of protein molecules spanning the entire thickness of the membrane provides a necessary link between the extracellular environment and the cell interior, which is consistent with the concept that drug activation of a membrane-bound receptor on the external surface of a cell can be directly translated into an intracellular response. Specific transmembrane proteins also provide important pathways for the uptake and extrusion of drugs.
Passive Diffusion
Simple diffusion
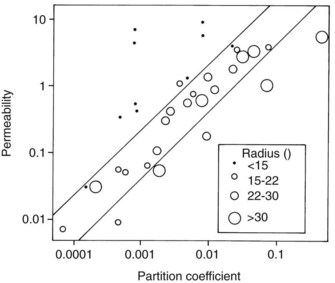
Studies by Overton and Meyer more than a century ago showed that the cell membrane acts for the most part as a lipoid barrier. As shown by Collander (Figure 2-3), the rate of transfer of nonelectrolytes across a membrane is directly proportional to the lipid/water partition coefficient. (The partition coefficient is a measure of the relative solubility of an agent in a fat solvent, such as olive oil or octanol, versus its solubility in water.) A drug with a high partition coefficient (i.e., a lipophilic drug) readily enters the lipid phase of the membrane and passes down its concentration gradient to the aqueous phase on the other side. More molecules are then free to enter the membrane and continue the transfer process. With poorly lipid-soluble compounds, however, few molecules enter the membrane per unit of time, and the rate of passage is depressed.
The absence of an ionic charge is one major factor favoring lipid solubility. Drugs with a fixed charge, such as drugs containing a quaternary nitrogen atom, permeate membranes slowly if at all. The reason for the relative solubility of nonionized molecules in lipids relates to their exclusion from polar media. Simple ions and charged molecules are stabilized in water by the hydration shells that surround them, a consequence of the tendency of charged species to orient polar molecules. This process excludes nonpolar substances, and the resulting segregation causes them to coalesce in a manner analogous to the formation of oil droplets on the surface of water. The term hydrophobic bonding, introduced in Chapter 1, refers to the tendency for water-insoluble molecules to be drawn together; this behavior is responsible for the preferential tendency of lipid-soluble drugs to penetrate cell membranes by way of the lipid components. Ionized compounds are so stabilized by their interaction with water that movement into a lipid phase is markedly restricted. Many therapeutic agents are weak electrolytes; depending on the pH of their aqueous environment, they can exist in ionized and neutral forms. Because charged molecules penetrate membranes with considerable difficulty, the rate of movement of these drugs is governed by the partition coefficient of the neutral species and the degree of ionization. As illustrated in Figure 2-4, acidic conditions favor the transport of weak acids, and the opposite holds true for basic compounds.
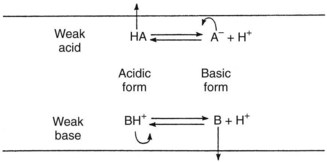
Figure 2-3 shows that lipid solubility is not the only factor influencing the simple diffusion of uncharged drugs across cell membranes; molecular size is also important. Water, glycerol, and some other small molecules permeate much more readily than would be predicted from their respective partition coefficients. Figure 2-3 also shows that some large organic molecules diffuse more slowly than expected. Nonelectrolytes containing numerous hydrophobic groups are often so insoluble in water that their transit across the lipid/water interface may be retarded despite a favorable partition coefficient.26 This finding suggests that some degree of water solubility is necessary for the passive diffusion of drugs across membranes. No matter how lipid soluble an agent is, it will never cross a membrane if it cannot first dissolve in the extracellular fluid and be carried to the structure. Benzocaine, an active local anesthetic when applied directly to nerves, is ineffective after injection because its water insolubility precludes significant diffusion away from the administration site and toward its locus of action within the neuronal membrane. When inside the membrane, a drug with an extremely high partition coefficient may be so soluble in the lipid phase that it has little tendency, despite moderate solubility in water, to diffuse out of the membrane down its concentration gradient.38 A review of human clinical data involving more than 2400 compounds suggests that simple diffusion will be poor if a drug has two or more of the following characteristics: (1) more than five H-bonding donor groups, (2) more than five H-bonding acceptor groups, (3) more than 10 N and O atoms, (4) a molecular weight greater than 500 Da, and (5) a partition coefficient greater than 10,000 : 1.28
Facilitated diffusion
Water, small electrolytes, and hydrophilic molecules of biologic importance generally move across plasma membranes much more readily than would be predicted by simple diffusion. In these instances, transmembrane proteins that circumvent the lipid bilayer facilitate diffusion. The simplest mechanism involves a transmembrane pore, such as aquaporin 1. Discovered in 1991, aquaporin 1 is a 28-kDa polypeptide that forms a 3-Å channel through which water can enter or leave cells. More than 10 aquaporins have been discovered in mammalian tissues and are especially prominent in cells and organs involved with the transcellular movement of water: kidneys, capillaries, secretory glands, red blood cells, choroid plexus, brain glia, eyes, and lungs.1,24 Some aquaporins are selective for water only, increasing its membrane permeability by a factor of 10 to 100; others permit the passage of glycerol and several other molecules in addition to water.
The movement of specific ions (e.g., Na+, K+, and Ca++) across the cell membrane is facilitated by the presence of transmembrane channels, such as the nicotinic receptor described in Figure 1-2 and the Na+ channel illustrated in Figure 16-4. The opening of these gated channels (in contrast to porins, which are always open) is regulated by the electric potential across the membrane or by the presence of specific ligands, such as neurotransmitters. When a channel is open, passive diffusion of an ion capable of traversing it depends on the electric potential across the membrane and the ion’s own chemical gradient. Boosting the electrochemical gradient by manipulating the voltage across the cell membrane is an effective method of increasing ionic flow. Even in the absence of specific ion channels, the transport of fixed ions and weak electrolytes across tissue barriers can be facilitated by the appropriate use of electric current (as in iontophoresis, discussed subsequently).
Active Transport
Approximately 49 ABC transporters hydrolyze adenosine triphosphate (ATP) to provide the energy directly needed for molecular transport and are referred to as primary active transporters. P-glycoprotein (P for altered permeability), also known as multidrug resistance protein-1 (MDR-1) and given the designation ABCB1 by the Human Gene Nomenclature Committee, is the most extensively researched representative. Originally identified in 1976 for its ability to expel numerous antineoplastic drugs from mutated cells that overexpress it, P-glycoprotein is a 170-kDa glycoprotein composed of two subunits in a head-to-tail arrangement (Figure 2-5).44,48 Each subunit contains a transmembrane domain of six α-helices that span the plasma membrane and help form the pump itself, and a nucleotide-binding domain (also known as the ABC cassette) that hydrolyzes ATP to power the transport. Many ABC transporters are referred to as half transporters because they consist of only a single subunit and must dimerize to create the active pump. P-glycoprotein preferentially promotes the cellular extrusion of large (300 Da to 2000 Da) hydrophobic substances and neutral or positively charged amphiphilic molecules. Transported drugs include numerous anticancer agents (e.g., doxorubicin, vinblastine, and paclitaxel), antiviral compounds (e.g., ritonavir), Ca++-channel blockers (e.g., diltiazem), digoxin, antibiotic and antifungal drugs (e.g., erythromycin and ketoconazole), hormones (e.g., testosterone), and immunosuppressants (e.g., cyclosporine).
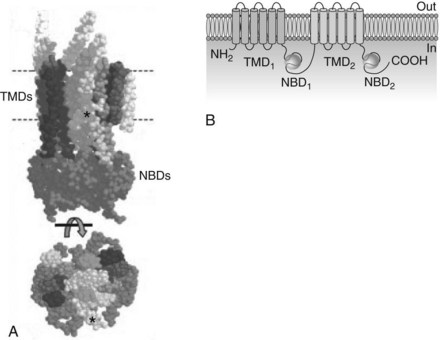
Drug binding occurs within the plasma membrane near the cytoplasmic surface, limiting transport to drugs with good lipid solubility or sufficient length to reach the active site. P-glycoprotein is expressed in various cells, but the highest concentrations are located in intestinal epithelial cells; renal proximal tubular cells; canalicular membranes of hepatocytes; the capillary endothelium of the brain, choroid plexus, testes, and placenta; placental trophoblasts; adrenocortical cells; and stem cells.30 Other ABC transporters important in pharmacokinetics include the multidrug resistance-associated protein (MRP) family. Collectively, the MRP transporters are also widespread and involved in the vectorial (one-way) movement of drugs and other xenobiotics. In contrast to P-glycoprotein, the MRP transporters pump amphiphilic molecules with at least one negative charge. These substrates include bile salts, nucleotide analogues, and conjugates of glutathione, glucuronic acid, and sulfate.
Organic anion transporters (OATs) and organic anion–transporting polypeptides (OATPs) are important families of SLC transporters involved in pharmacokinetics.35 As a group, they promote the cellular uptake of acidic drugs into the liver, kidney, intestine, lung, and brain, and their excretion via the bile and urine. An analogous family of organic cation transporters (OCTs) provides similar handling of positively charged drugs.
Endocytosis and Exocytosis
Endocytosis usually begins with the binding of a compound, usually a macromolecule, to be absorbed by its receptor on the membrane surface. Two good examples are the attachment of low-density lipoprotein (LDL) and insulin to their respective receptors. With time, the bound agent-receptor complex is concentrated in an indentation of the membrane called a coated pit. (This migration also occurs spontaneously with the LDL receptor.) Clathrin, a cytoplasmic protein that attaches to the internal surface of the plasma membrane, serves to capture the receptors within the pit while excluding other surface proteins.49 Internal rearrangement of its structure deepens the pit, forming a coated bud. A second protein, termed dynamin, is believed to congregate around the collar of the invaginated bud and initiate separation from the membrane. When released, the vesicle loses its clathrin coat and fuses with an organelle called the endosome. Some of the captured contents, such as LDL receptors, are recycled back to the plasma membrane by transport vesicles; the remainder undergo lysosomal processing and release into the cytoplasm.
ABSORPTION
Oral Ingestion
Influence of pH
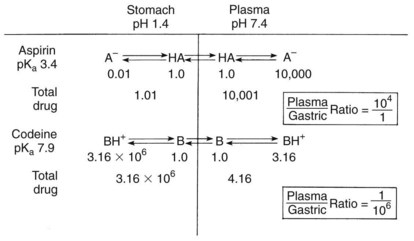
As previously discussed, absorption is favored when the drug ingested is lipid soluble. For weak electrolytes, the pH of the surrounding medium affects the degree of ionization and drug absorption. Because the H+ concentrations of the stomach and small intestine diverge widely, the two structures seem to be qualitatively dissimilar in their respective patterns of drug absorption. Figure 2-6 illustrates this difference and its effect on the previously commonly used analgesic combination of aspirin plus codeine. Aspirin is an organic acid with a pKa (negative log of the dissociation constant) of 3.49. In gastric juice (pH 1 to 3), aspirin remains largely nonionized, and its passage across the stomach mucosa and into the bloodstream is favored. The plasma has a pH of 7.4, however. On entering this environment, the aspirin becomes ionized to such an extent that return of the drug to the gastrointestinal tract is prevented by the low lipid solubility of the anionic species. When equilibrium is established, the concentration of nonionized aspirin molecules on both sides of the membrane is the same, but the total amount of drug (ionized plus neutral forms) is much greater on the plasma side. The relative concentration of drug in each compartment can be calculated with the Henderson-Hasselbalch equation, as follows:
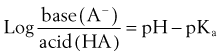
Mucosal surface area
A second major difference between absorption in the stomach and absorption in the small intestine relates to the intraluminal surface areas involved in drug uptake. Aside from certain mucosal irregularities (rugae), the stomach lining approximates that of a smooth pouch with a thick mucous layer. The mucosa of the small intestine is uniquely adapted for absorption, however. Contributions by the folds of Kerckring, villi, and microvilli combine to increase the effective surface area 600-fold. Assuming a small intestine 280 cm in length and 4 cm in diameter, approximately 200 m2 are available for drug absorption. The surface/volume ratio in the small intestine is so great that drugs ionized even to the extent of 99% may still be effectively absorbed. Many studies have shown that acidic drugs with a pKa greater than 3.0 and basic compounds with a pKa less than 8.0 readily pass from the intestinal fluid into the plasma.19 Although pH considerations favor the gastric absorption of aspirin, as much as 90% of the drug when given in tablet form is actually absorbed from the small intestine in vivo. Experimentally, nonelectrolytes such as ethanol are also absorbed from the intestine many times faster than from the stomach.
Gastric emptying
Additional situations in which food enhances drug uptake have been reviewed.33 Nevertheless, because gastric emptying is often a limiting factor in the rate of drug absorption, many unrelated drugs exhibit latency periods (the lag phase between oral ingestion and onset of drug effect) of a similar magnitude.
Influence of dosage form
The first step in the dissolution process is the disintegration of the tablet (or the capsule and its granules) to yield the primary drug particles. Various excipients are usually included in solid drug preparations to promote disintegration and particle dispersion. If disintegration is impaired, drug absorption is depressed accordingly. The dissolution of drug particles occurs by a diffusion-limited mechanism. The diffusion layer of solvent surrounding each particle becomes saturated very quickly with drug molecules escaping from the solid. Because saturation of the diffusion layer occurs far more rapidly than does diffusion from it into the bulk solution, the entire process proceeds no faster than the rate of drug diffusion. Several methods can be used, however, to accelerate the dissolution rate. Because the total surface area of the particles determines the area available for diffusion, reducing the mean particle size through the process of micronization promotes solubilization. A decrease in particle size of 85% with a compensating increase in particle number doubles the rate of dissolution.27 Another useful approach is to manufacture drugs in the form of water-soluble salts. The concentration of drug in the dissolution layer is enhanced (often by many times), and the rate of diffusion is increased.
The dissolution process may be considered rate limiting whenever a drug solution produces a systemic effect faster than a solid formulation of the same agent does. Sometimes discrepancies in absorption between dosage forms are of such magnitude that clinical differences are noted. With aspirin, the concentration of drug in the plasma 30 minutes after administration can be twice as high for a solution as for a solid tablet.27 Although it is unclear whether this difference results solely from drug dissolution or from other factors, such as the more rapid gastric emptying typical of liquids, dissolution is probably at least partially responsible.
The influence of dosage form on drug absorption is often taken advantage of by drug manufacturers. Some drugs (e.g., erythromycin) are unstable at a low pH, and others (e.g., ammonium chloride) are irritating to the gastric mucosa. To avoid release of these drugs within the stomach, they are often prepared in the form of enteric-coated tablets. An enteric coat consists of a film of shellac or some polymeric substitute. The covering is insoluble under acidic conditions, but does break down to permit tablet disintegration in the more alkaline environment of the small intestine. Although these preparations are often beneficial, their usefulness nevertheless is negatively affected by an increased variability in patient response. Because drug absorption cannot begin until the tablet passes into the duodenum, the time required for gastric transit becomes an important variable. The passage of a single insoluble tablet from the stomach into the intestine is a random event that can take several minutes to more than 6 hours.15
The sensitivity of gastrointestinal absorption to variations in drug formulation is best exemplified by the concern over bioavailability. In many instances, chemically identical drugs have proved in the past to be biologically nonequivalent. In one study of tetracycline hydrochloride, nine preparations of different manufacture were compared with an aqueous solution of the same drug.29 Although seven brands produced blood concentrations ranging from 70% to 100% of the reference solution, two products exhibited relative bioavailabilities of only 20% to 30%. Differences in bioavailability are most likely to be clinically important with drugs that are poorly absorbed, have low margins of safety, and are inactivated by capacity-limited processes. Since 1977, federal law has required that bioequivalence testing be performed on all new drugs, and the FDA has mandated such testing of existing products for which a problem of nonequivalence is known to exist. Bioavailability considerations related to drug selection are considered further in Chapter 55.
Active transport
Active transport mechanisms can also inhibit drug absorption.30 P-glycoprotein is highly expressed along the luminal surface of intestinal epithelial cells, where it exports xenobiotics that would otherwise be absorbed. This function is in concert with the “chemoimmunity defensive” role P-glycoprotein plays in protecting cells from exposure to potentially toxic compounds.48 Although P-glycoprotein may delay the absorption of many drugs and prevent altogether the uptake of pharmaceuticals of low absorptive potential, it is probably of minor significance regarding the extent of absorption of most drugs intended for oral use, whose concentrations in the chyme are sufficient to overwhelm the capacity of P-glycoprotein to export them.47 Figure 2-7 depicts the active transport of drugs into and out of intestinal cells and at other important sites.

Other enteral routes
The oral and rectal mucosae are occasionally used as sites of drug absorption. Sublingual administration, in which a tablet or troche is allowed to dissolve completely in the oral cavity, takes advantage of the permeability of the oral epithelium and is the preferred route for a few potent lipophilic drugs, such as nitroglycerin and oxytocin. The oral and intestinal mucosal layers do not differ qualitatively as absorbing surfaces, and comparable absorption has been shown for many agents.6 One reason for selecting the sublingual route is to avoid drug destruction. Because gastric acid and intestinal and hepatic enzymes are bypassed, sublingual absorption can be more efficient overall for certain drugs than is intestinal uptake. The onset of drug effect may also be quicker than with oral ingestion.
Inhalation
Therapeutic use of aerosols is not widespread, but some emergency medications are prepared in this form. Because the onset of effect is extremely rapid after inhalation of an aerosol drug, this route can provide a means of quick self-medication for individuals in danger of acute allergic reactions to venoms or drugs. Epinephrine is one such emergency agent that is marketed as an aerosol. Many respiratory drugs are also prepared in aerosol form because they are highly effective by this route while minimizing systemic exposure. The rapidity and efficiency of alveolar membrane absorption can occasionally pose problems for therapy, however, as illustrated by the use of pressurized aerosols containing isoproterenol. Although 97% of an isoproterenol spray is swallowed under normal conditions and inactivated by various enzymes, overmedication can produce toxic effects. Data gathered over a 7-year period in the United Kingdom suggested that the undisciplined use of these preparations increased mortality in asthmatic patients. Restriction of over-the-counter sales and warnings to physicians were accompanied by a decline in mortality.21 Findings such as these reflect the hazards of aerosols when abused and provide a caveat for uncontrolled self-medication with any potentially dangerous drug. Concern over aerosols is also related to questions of toxicology, such as the absorption of heavy metal dusts by industrial workers.
Parenteral Injection
Stay updated, free dental videos. Join our Telegram channel
VIDEdental - Online dental courses


