Autoimmune disease
Key Points
The normal immune response and tests are summarized in Chapter 19.
Many diseases are immunologically mediated and this is especially notable in allergies and autoimmunity. A genetically determined susceptibility to immunologically mediated diseases is often suggested by a positive family history, and by significant associations with certain major histocompatibility complex (MHC) histocompatibility (human lymphocyte antigen, or HLA) antigens. The genes for histocompatibility antigens are identified by a letter (A–D) and a number (e.g. HLA-D2). This chapter discusses autoimmune disorders – mainly those that affect multiple tissues; organ-specific disorders are discussed in the appropriate chapters (Table 18.1). Autoimmunity is an immune response that destroys normal body tissues. There are more than 80 different autoimmune disorders and they may result in: destruction of body tissue; abnormal growth of or changes in organ function.
Table 18.1
| Non-organ-specific autoantibodies: the connective tissue diseasesa | Organ or cell-specific autoantibodiesb |
| Raynaud disease | Autoimmune haemolytic anaemia |
| Lupus erythematosus | Chronic atrophic gastritis (pernicious anaemia)c |
| Rheumatoid arthritis | Hashimoto thyroiditis |
| Sjögren syndrome | Idiopathic Addison disease |
| Scleroderma | Idiopathic hypoparathyroidism |
| Dermatomyositis | Idiopathic thrombocytopenic purpura |
| Mixed connective tissue disease | Myasthenia gravis |
| Antiphospholipid syndrome | Pemphigoid |
| Pemphigus vulgaris |
aProbably mediated by immune complex reactions: discussed in this chapter.
The causes of autoimmune disease are unclear but heredity appears to play a role; several polymorphic genes (i.e. HLA-DRB1, tumour necrosis factor [TNF] and PTPN22 [protein tyrosine phosphatase non-receptor 22]) influence the susceptibility to different autoimmune diseases. Regulatory T cells (Treg) fail in their function. Many organ-specific autoimmune diseases are associated with HLA-B8 and DR3, but the association is not sufficiently strong to help in the diagnosis. Rheumatoid arthritis, by contrast, is associated with HLA-DR4.
General aspects
Autoimmune diseases are caused by antibodies and T cells directed against self, produced when immune system recognition fails or malfunctions. Autoimmune diseases may be initiated by viruses, drugs, chemicals or other factors, and affect the immune system at several levels. In patients with systemic lupus erythematosus, for instance, B cells are hyperactive while suppressor cells are underactive; it is not clear which defect comes first. Moreover, production of interleukin-2 (IL-2) is low, while levels of interferon-gamma (IFN-γ) are high. Patients with rheumatoid arthritis, who have defective suppressor T cells, continue to make antibodies to common viruses, a response that would normally cease after about 14 days. Some autoantibodies are directed against specific antigens and cause localized disease (e.g. autoantibodies to desmoglein in epithelial intercellular cement, which cause pemphigus; autoantibodies to red blood cells, which cause anaemia; autoantibodies to pancreatic islet cells, which contribute to juvenile diabetes; and autoantibodies to acetylcholine receptors, which are present in myasthenia gravis). Polyglandular autoimmune diseases are examples of autoimmune attack on many organs.
Where autoantibodies are directed against several types of cell and cellular components, including nuclear components such as deoxyribonucleic acid (DNA), ribonucleic acid (RNA) or proteins, then generalized disease, such as systemic lupus erythematosus, may result (see Table 18.1).
Clinical features
Autoimmune diseases share common features (Table 18.2).
Table 18.2
Typical features of autoimmune diseases
| Clinical features | Laboratory features |
| Female predisposition | Hypergammaglobulinaemia |
| Family history often positive | Autoantibodies specific to tissue under attack sometimes present in the circulation. Circulating autoantibodies often detectable in apparently unaffected relatives. Multiple autoantibodies often detectable but frequently without clinical effect |
| Response to immunosuppressive treatment in many | Immunoglobulin and complement deposits sometimes detectable by immunofluorescence microscopy at sites of tissue damage (e.g. damaged blood vessels) |
| Patients especially liable to develop further autoimmune diseases | Associations with HLA-B8 and DR3 in some |
General management
The blood picture may show results of autoantibodies against haematological factors (e.g. haemolytic anaemia, thrombocytopenia or leukopenia). Alternatively, there may be leukocytosis associated with inflammatory processes. The erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are is often raised. Serum protein levels are also typically increased because of immunoglobulin overproduction. Specific tests for autoimmune disease include assays for serum autoantibodies (see Ch. 2 and Table 18.3). Specific autoantibodies can be identified by special tests but often an autoantibody profile is carried out (this includes the most common abnormalities – namely, rheumatoid and antinuclear factors and thyroid microsomal, gastric parietal cell, mitochondrial, smooth muscle and reticulin antibodies). Serum complement levels may show the complement consumed when the cascade is activated by antigen/antibody reactions or other triggering factors, with complement levels (CH50) often depressed. Biopsy and tissue immunostaining may show immunoglobulin or complement deposits. Vasculitis with deposits of immunoglobulins and complement in the damaged vessel walls strongly suggests immune complex injury.
Table 18.3
Significance of the more common antinuclear antibodies related to connective tissue diseasesa
< ?comst?>
| Autoantibodies | Main associations |
| Anti-Jo 1 (GAD65; histidine tRNA ligase) | Polymyositis, dermatomyositis, anti-synthetase syndrome |
| Anti-Ku | Polymyositis/scleroderma (PM/Scl) overlap syndrome |
| Anti- double-stranded (anti-ds) DNA | Antibody with the highest specificity for systemic lupus erythematosus (SLE) and found in most patients |
| Anti-extractable nuclear antigen (anti-ENA antibodies) | SLE |
| Anti-Sm antibody (snRNP) | |
| Anti-extractable nuclear antigen (anti-ENA antibodies) directed against RNP | Sjögren syndrome |
| Anti-Ro (Robair)=SS-A | |
| Anti-extractable nuclear antigen (anti-ENA antibodies) directed against RNP | Sjögren syndrome |
| Anti-La (Lattimer)=SS-B | |
| Anti-extractable nuclear antigen (anti-ENA antibodies) | Mixed connective tissue disease (MCTD), SLE |
| Anti-snRNP70 | |
| Anti-PM/Scl (anti-exosome) | PM/Scl overlap syndrome |
| Anti-histone | Drug-induced lupus, SLE |
| Anti-Scl 70 | |
| Anti-topoisomerase | Scleroderma |
| Anti-centromere | CREST syndrome (calcium deposits; Raynaud phenomenon; oesophageal dysfunction; sclerodactyly; and telangiectasias) |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
Treatments for autoimmune diseases include mainly anti-inflammatory and immunosuppressive drugs (including biologics) or plasmapheresis (removal of the antibodies).
The Connective Tissue Diseases
The connective tissue diseases (collagen diseases) are autoimmune diseases that share, to a variable degree, common features, such as: multiple autoantibodies, often against nuclear components (antinuclear antibodies [ANAs]; see Table 18.3); immune complex reactions; associations with Raynaud phenomenon and Sjögren syndrome; responsiveness to immunosuppressives; and malignancy as a possible late complication.
Antinuclear antibodies
ANAs are found in a number of different autoimmune diseases (such as systemic lupus erythematosus, Sjögren syndrome, rheumatoid arthritis, polymyositis, scleroderma, juvenile diabetes mellitus, Addison disease, vitiligo, pernicious anaemia, glomerulonephritis); infections (viral or bacterial, mainly); lung diseases (primary pulmonary fibrosis, pulmonary hypertension); gastrointestinal diseases (ulcerative colitis, Crohn disease, primary biliary cirrhosis, alcoholic liver disease); hormonal diseases (Hashimoto autoimmune thyroiditis, Graves disease); blood diseases (idiopathic thrombocytopenic purpura, haemolytic anaemia), cancers (melanoma, breast, lung, kidney, ovarian and others); and skin diseases (psoriasis, pemphigus). They are also found in those people with a family history of rheumatic diseases and in older patients, and in association with some drugs, including procainamide, hydralazine and phenytoin. Since ANAs appear in several types of disease, they have a low specificity.
ANAs can present different ‘patterns’ on histology, depending on the staining of the cell nucleus in the laboratory – homogeneous or diffuse; speckled; nucleolar; and peripheral or rim – but these patterns are not specific for any particular illness, though (for example) the nucleolar pattern is more commonly seen in scleroderma. ANAs can be found in approximately 5% of the normal population, usually in low titres; these people rarely have disease. ANA titres of 1:40 or below are considered negative. Titres lower than 1:80 are unlikely to be significant. Higher titres may be indicative of disease, but even these levels are often insignificant in patients over 60 years of age. Nevertheless, the sensitivity and simplicity of an ANA test makes it popular as a screen for lupus in particular, since most affected people (more than 95%) test positive, and thus a negative ANA test can be helpful in excluding lupus. Only about 11–13% of people with a positive ANA test do have lupus and up to 15% of completely healthy people have a positive ANA test. Thus a positive ANA test does not necessarily mean a diagnosis of lupus or, indeed, any autoimmune disease. Tests for ANA sub-serologies (including antibodies to double-stranded DNA, Smith, RNP, SSA, SSB, Scl-70 and centromere) are usually negative if ANA is negative. Exceptions include anti-Jo1, which can be positive in some forms of myositis, or occasionally anti-SSA, in the setting of lupus or Sjögren syndrome. Broad testing of autoantibodies should be avoided; instead, the choice of autoantibodies should be guided by the specific disease under consideration.
Raynaud Disease and Raynaud Phenomenon
Raynaud phenomenon manifests as recurrent vasospasm of the fingers and toes – usually in response to stress or cold exposure. Raynaud phenomenon is characterized by skin arteries that over-respond to cold, contracting briefly and limiting blood flow (vasospasm), so that the skin first turns white, then blue and finally red, as the arteries relax and blood flows again. Women are mainly affected.
Primary Raynaud disease (idiopathic Raynaud disease) is characterized by vasospasm alone and has no obvious cause, but 10–30% eventually progress to secondary Raynaud disease. Older patients and males often have vasospastic symptoms that predate systemic disease by as many as 20 years. Young female patients who have had Raynaud phenomenon alone for more than 2 years and have not developed any additional manifestations are, however, at low risk for developing autoimmune disease.
Secondary Raynaud disease is a term used when vasospasm is associated with another condition – most commonly an autoimmune disease, and usually progressive systemic sclerosis and mixed connective-tissue disease. Raynaud phenomenon has also been described with disorders such as cryoglobulins, vibration injury, polyvinyl chloride (PVC) exposure and drugs (e.g. some cardiovascular drugs and ergotamine). In older patients with recent-onset Raynaud phenomenon and no obvious underlying cause, malignancy must also be excluded.
Clinical features
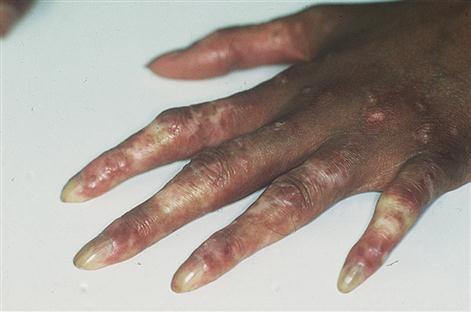
Symptoms include changes in skin colour (white to blue to red) and coldness (Fig. 18.1). Both hands and feet are usually affected. Raynaud phenomenon can sometimes attack other areas too, such as the nose and ears. It is common for the affected area to feel numb or prickly. Primary Raynaud disease usually affects both hands and both feet, while secondary Raynaud phenomenon may affect either both hands or both feet only.
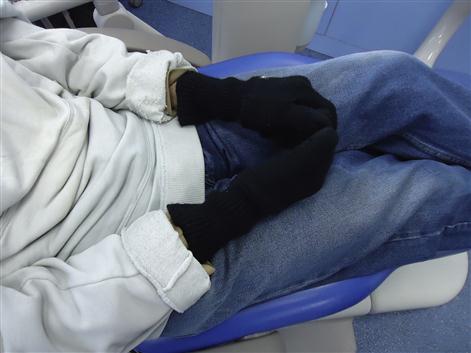
The condition tends to be slowly progressive, with more frequent and prolonged episodes of spasm. Ischaemia can cause atrophy of the fat pads or ulceration at the tips of the fingers or toes, sometimes with the loss of more tissue (Fig. 18.2).
General management
Diagnosis is made from the fact that the attacks are triggered by exposure to cold and/or stress, symmetric bilateral involvement, an absence of necrosis, no detectable underlying cause, normal capillaroscopy findings, normal laboratory findings for inflammation, and an absence of antinuclear factors.
People suffering from Raynaud phenomenon should stop smoking (nicotine is vasoconstricting), protect themselves from cold, keep all their body warm – not just their extremities – and guard against cuts, bruises and other injuries to affected areas.
Primary Raynaud disease
Secondary Raynaud disease
Therapy should be tailored to the underlying disorder:
Pharmacological therapy is as for primary Raynaud disease.
Dental aspects
The dental environment should be kept at a comfortable temperature and humidity. Nifedipine may cause gingival swelling. Sjögren syndrome may be present.
Lupus Erythematosus
Lupus can be categorized as systemic lupus erythematosus (SLE), drug-induced SLE or discoid lupus erythematosus (DLE).
Systemic lupus erythematosus
General aspects
SLE is a potentially fatal disease characterized by autoantibodies to DNA (anti-dsDNA [double strand DNA] antibody is peculiar to SLE). The immune complex reactions, in particular vasculitis, result in multisystem disease. Lupus susceptibility relates to several genes, including HLA-DR3 and B8, HLA-DR2, complement C4 genes and a polymorphism of the T-cell receptor that has been associated with Ro (SS-A) antibodies.
Clinical features
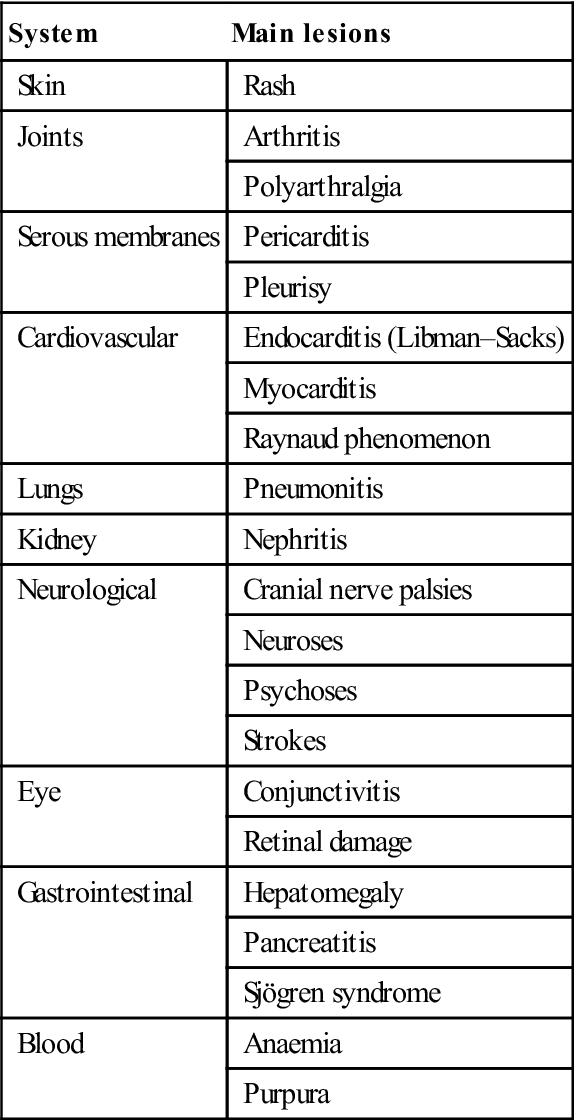
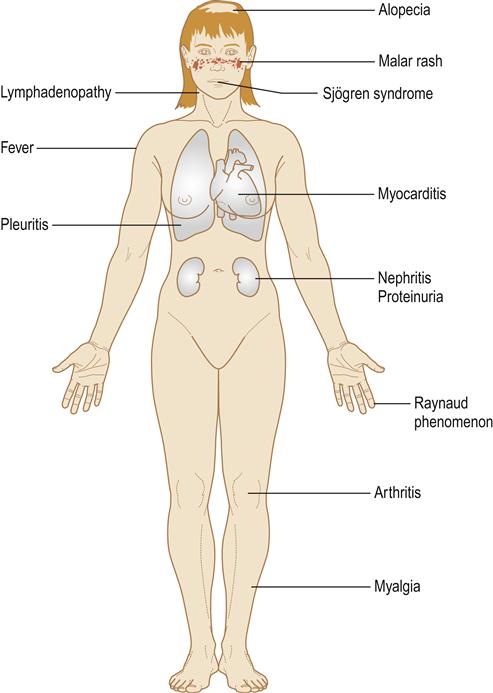
SLE can cause a variety of clinical manifestations, which differ greatly in severity, ranging from mild to significant and potentially fatal lesions. The onset may be acute, resembling an infection, or there may be vague symptoms that may progress over many years (Table 18.4). The acronym SOAPBRAIN may serve as an aide-mémoire (serositis; oral ulcers; arthritis; pericarditis; blood problems; renal; ANA; immune; neurological).
Table 18.4
Manifestations of systemic lupus erythematosus
< ?comst?>
| System | Main lesions |
| Skin | Rash |
| Joints | Arthritis |
| Polyarthralgia | |
| Serous membranes | Pericarditis |
| Pleurisy | |
| Cardiovascular | Endocarditis (Libman–Sacks) |
| Myocarditis | |
| Raynaud phenomenon | |
| Lungs | Pneumonitis |
| Kidney | Nephritis |
| Neurological | Cranial nerve palsies |
| Neuroses | |
| Psychoses | |
| Strokes | |
| Eye | Conjunctivitis |
| Retinal damage | |
| Gastrointestinal | Hepatomegaly |
| Pancreatitis | |
| Sjögren syndrome | |
| Blood | Anaemia |
| Purpura |
< ?comen?>< ?comst1?>
< ?comst1?>
< ?comen1?>
The classic picture of SLE is that of a young woman with fever, malaise, anaemia, joint pains, Raynaud phenomenon and a rash variable in character but erythematous, often with raised margins and scaling (Fig. 18.3). The well-known rash, with a butterfly pattern extending over both cheeks and bridge of the nose, is relatively uncommon and not specific to lupus erythematosus. Photosensitivity rashes are common. Renal involvement (proteinuria and haematuria) is frequently seen and often associated with hypertension. Relatively minor disturbances of mood, and depressive or hysterical behaviour, are common, while central nervous system (CNS) involvement is potentially fatal. Ocular lesions may affect 20–25%, and serositis can cause pleurisy or occasionally pericarditis and peritonitis (polyserositis) (Fig. 18.4).
Cardiac lesions typically cause myocarditis, which leads to cardiac failure; there is a higher risk of infarction, and a characteristic (Libman–Sacks) endocarditis can also develop. Murmurs are more often caused by anaemia.
Women who have SLE can pass antibodies across the placenta, which can cause fetal heart block (see Sjögren syndrome).
General management
The American College of Rheumatology diagnostic criteria for SLE are based on the presence of at least four of the following:
■ Serositis (pleuritis or pericarditis)
■ Renal disorder (persistent proteinuria or cellular casts)
Initial screening includes ESR, autoantibodies (e.g. ANA; Box 18.1), a full blood count, liver and kidney function tests, syphilis screen (VDRL), lupus erythematosus cell test, urinalysis and blood chemistries. No single laboratory test can definitely prove or exclude SLE, but anti-Sm antibody or high-titre anti-native DNA antibody is an important indication (see Table 18.3); anti-dsDNA is peculiar to SLE and the titre of anti-DNA antibodies correlates well with disease activity. About 70% of patients have antibodies against double-stranded RNA (anti-dsRNA) or against hybrid RNA/DNA molecules. Other autoantibodies (e.g. anti-SS-A and anti-SS-B) may be helpful. About 40% of patients also have antibodies directed against phospholipid – and about 15% of patients have the antiphospholipid syndrome (APS; see later).
Biopsies of skin or kidney using immunofluorescent staining techniques can support the diagnosis of SLE.
Most patients appear to do well in the long term either with non-steroidal anti-inflammatory drugs (NSAIDs) or, if these are ineffective, on low doses of corticosteroids taken on alternate days. Other treatment options currently available include hydroxychloroquine and other immunosuppressive medications (e.g. azathioprine, ciclosporin, cyclophosphamide, mycophenolate or sirolimus), as well as a range of biological agents (abatacept, abetimus, anakinra, atacicept, belimumab, edratide, efalizumab, epratuzumab, infliximab, rituximab, rontalizumab, sifalimumab and tocilizumab). SLE has a 5-year survival rate of more than 90%.
Dental aspects
Oral lesions, which typically consist of erythematous areas, erosions or white patches, fairly symmetrically distributed and resembling lichen planus, may be seen in 10–20% of patients with SLE and are rarely an early feature. Slit-like ulcers may also be seen near the gingival margins. The best management of erosive lesions of SLE in the oral mucosa is uncertain but corticosteroids, often in high doses, may be the only effective treatment. Biopsies of oral lesions show irregular epithelial thinning and acanthosis, basement membrane thickening, liquefaction degeneration of the basal cell layer and an irregularly distributed chronic inflammatory infiltrate. Vasculitis is inconstant. Immunofluorescent staining shows lumpy deposits of immunoglobulins and complement perivascularly and along the basement membrane of lesions, and under normal skin or mucosa in up to 90% of patients with SLE. Sjögren syndrome is associated in up to 30%. Antimalarials, sometimes used to control SLE, can cause lichenoid oral lesions or occasionally oral hyperpigmentation.
Surgery may exacerbate SLE symptoms and, if elective, should therefore be postponed until lupus activity subsides. Hospitalization may be required for otherwise minor procedures and postoperative discharge may be delayed.
Tetracyclines may cause photosensitivity rashes while sulphonamides or penicillins may lead to deterioration in SLE. Bleeding tendencies are caused by thrombocytopenia; circulating anticoagulants rarely cause a bleeding tendency – rather they predispose to thrombosis. Other aspects to consider are corticosteroid or other immunosuppressive therapy (Ch. 35), renal disease (Ch. 12) and anaemia (Ch. 8).
Drug-induced lupus (Table 18.5) has some features similar to those of SLE, particularly pleuropericardial inflammation, fever, rash, arthritis and serological changes. The clinical and serological signs of the lupus erythematosus usually subside gradually after the offending drug is discontinued.
Table 18.5
Drugs associated with lupoid reactions
| Drugs having a proven association with LE | Drugs having a possible association with LE |
| Chlorpromazine | Beta-blockers |
| Hydralazine | Captopril |
| Isoniazid | Carbamazepine |
| Methyldopa | Cimetidine |
| Procainamide | Etanercept |
| Ethosuximide | |
| Methimazole | |
| Phenazine | |
| Phenytoin (diphenylhydantoin) | |
| Quinidine |
Discoid lupus erythematosus
General aspects
Discoid lupus erythematosus (DLE) is essentially a mucocutaneous disease in which the rashes may be indistinguishable from those of SLE, but serological abnormalities are typically absent or minor, there are no serious systemic effects and it only rarely progresses to SLE.
Clinical features
DLE is characterized by a rash, usually on the face or other sun-exposed areas, or by mucosal lesions. The skin lesions are patchy, crusty, sharply defined plaques that may scar. DLE may cause patchy, bald areas on the scalp, and hypopigmentation or hyperpigmentation in older lesions. Arthralgia is a frequent complaint.
General management
Lesional biopsy will usually confirm the diagnosis. Topical and intralesional corticosteroids are usually effective for localized lesions; antimalarial drugs may be needed for more generalized lesions.
Dental aspects
Oral mucosal lesions can be a feature of DLE and may also simulate lichen planus, but the lesions are less often symmetrically distributed and the pattern of striae is typically less well defined or conspicuous. Nevertheless, differentiation can be difficult and biopsy is essential. Even this may not be diagnostic and, occasionally, histological appearances that are intermediate between those of lichen planus and lupus erythematosus are seen. Management is as for the oral lesions of SLE but DLE may have some malignant potential.
Relapsing Polychondritis
Relapsing polychondritis (RP) is a severe, episodic and progressive inflammatory condition associated with antibodies to cartilage- specific collagen types II, IX and XI. RP affects cartilaginous structures, predominantly those of the ears, nose and laryngotracheobronchial tree. Other affected structures may include the eyes, cardiovascular system, peripheral joints, skin, middle and inner ear, and CNS. Aphthous-like oral ulceration has been described and the MAGIC variant of Behçet syndrome is a differential diagnosis. Management is with systemic corticosteroids.


